In this demo, we will be analyzing Visium data on a human colorectal cancer biopsy from de Oliveira et al. (2025). Rather than recapitulating all possible analyses, our goal is to highlight those that might be of particular interest in the context of these data.
# retrieve dataset from OSF repoid<-"Visium_HumanColon_Oliveira"pa<-OSTA.data_load(id)dir.create(td<-tempfile())unzip(pa, exdir=td)# read into 'SpatialExperiment'obj<-TENxVisium( spacerangerOut=file.path(td, "outs"), format="h5", images="lowres")(spe<-import(obj))
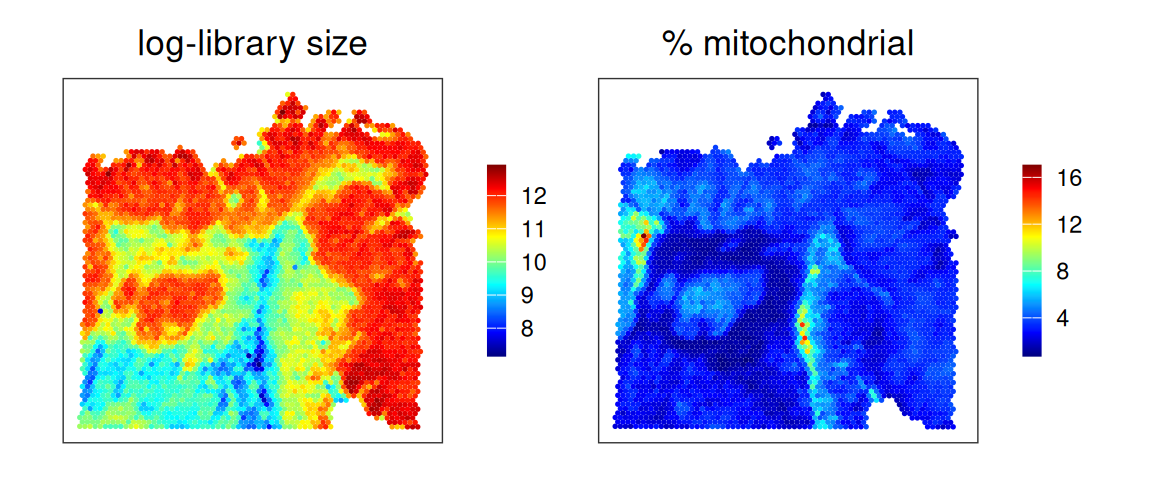
15.3 Quality control
Code
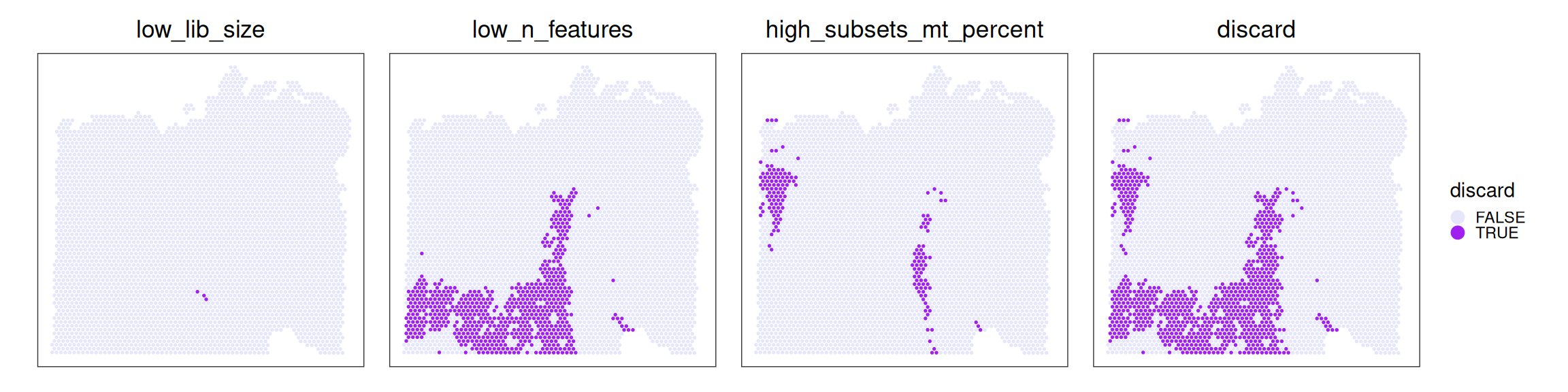
# use gene symbols as feature namesrownames(spe)<-make.unique(rowData(spe)$Symbol)# add per-cell quality control metrics & determine # outliers via thresholding on MAD from the mediansub<-list(mt=grep("^MT-", rownames(spe)))spe<-quickRnaQc.se(spe, subsets=sub)spe$discard<-!spe$keepspe$log_sum<-log1p(spe$sum)spe$mt_prop<-spe$subset.proportion.mt
Code
# tabulate # & % of cells that'd be # discarded for different reasonsths<-metadata(spe)$qc$thresholdsols<-data.frame( low_sum=spe$sum<ths$sum, low_detected=spe$detected<ths$detected, high_mt_prop=spe$mt_prop>ths$subset.proportion, discard=spe$discard)data.frame( check.names=FALSE, `#`=apply(ols, 2, sum), `%`=round(100*apply(ols, 2, mean), 2))
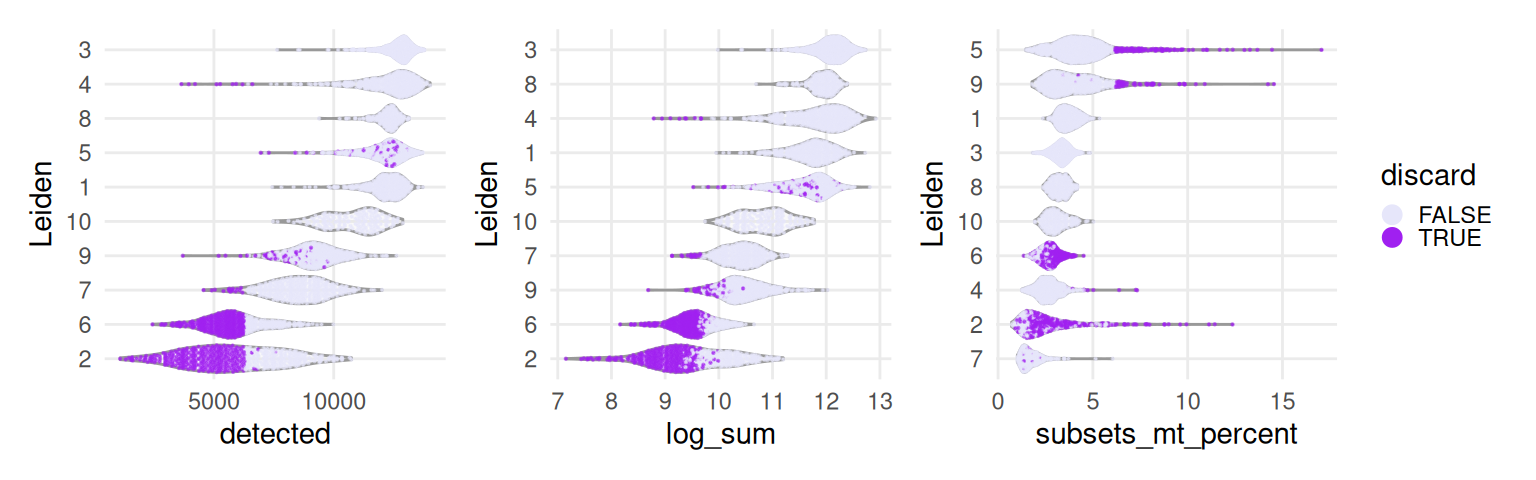
It seems like low-quality spots are highly spatially organized, so that might encourage us to not remove them, for now. We will see further below how the quality control metrics used here, and spots deemed to be discarded, are distributed across (transcription-based) clusters.
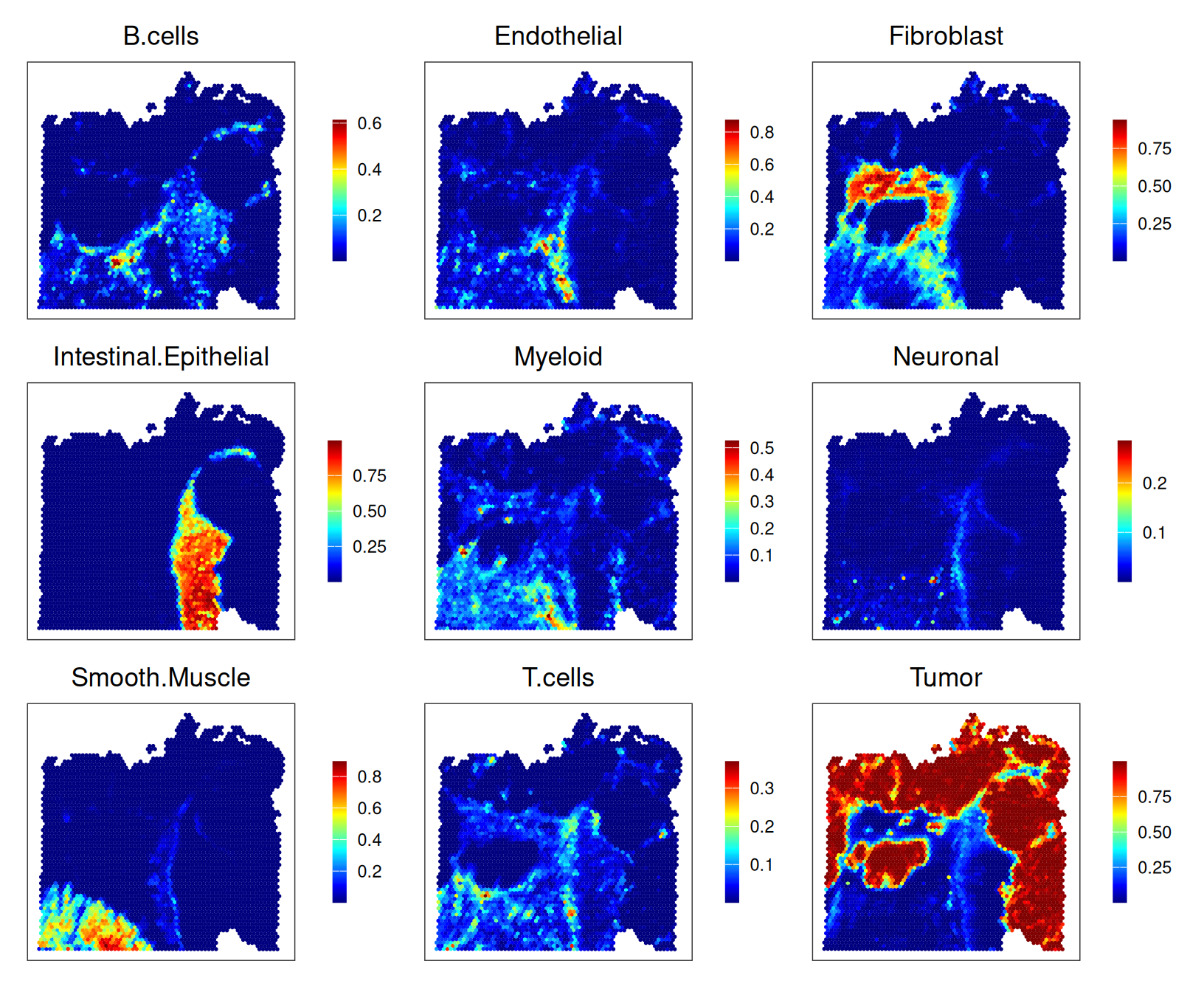
In a complementary approach, we deconvolve spot measurements using (annotated) reference single-cell data provided by the authors. Let’s first retrieve these data, alongside corresponding cell metadata, which includes low- (Level1) and high-resolution (Level2) annotations into 10 and 32 subpopulations, respectively.
Next, we perform deconvolution with spacexr’s (RCTD) (Cable et al. 2022). By default, runRctd()’s rctd_mode="doublet", i.e., at most two subpopulations are fit per pixel; here, we set rctd_mode="full" in order to allow for an arbitrary number of subpopulations to be fit instead.
Here, we filter the reference data to contain only cells from the same patient, and downsample to retain a limited number of cells per subpopulation. This is not strictly necessary, assuming that clusters are transcriptionally stable across patients, but helps with reducing runtime and memory consumption here.
Code
# prep reference data (Chromium);# subset cells from same patient.sce<-sce[, grepl("P2", sce$Patient)]# downsample to at most 2,000 cells per clustercs<-split(seq_len(ncol(.sce)), .sce$Level1)cs<-lapply(cs, \(.)sample(., min(length(.), 2e3))).sce<-.sce[, unlist(cs)]# run 'RCTD' deconvolutionrctd_data<-createRctd(spe, .sce, cell_type_col="Level1")(res<-runRctd(rctd_data, max_cores=th, rctd_mode="full"))
Weights inferred by RCTD should be normalized such that proportions of cell types sum to 1 in each spot:
Code
# scale weights such that they sum to 1ws<-assay(res)ws<-sweep(ws, 2, colSums(ws), `/`)# add proportion estimates as metadataws<-data.frame(t(as.matrix(ws)))colData(spe)[names(ws)]<-ws[colnames(spe), ]
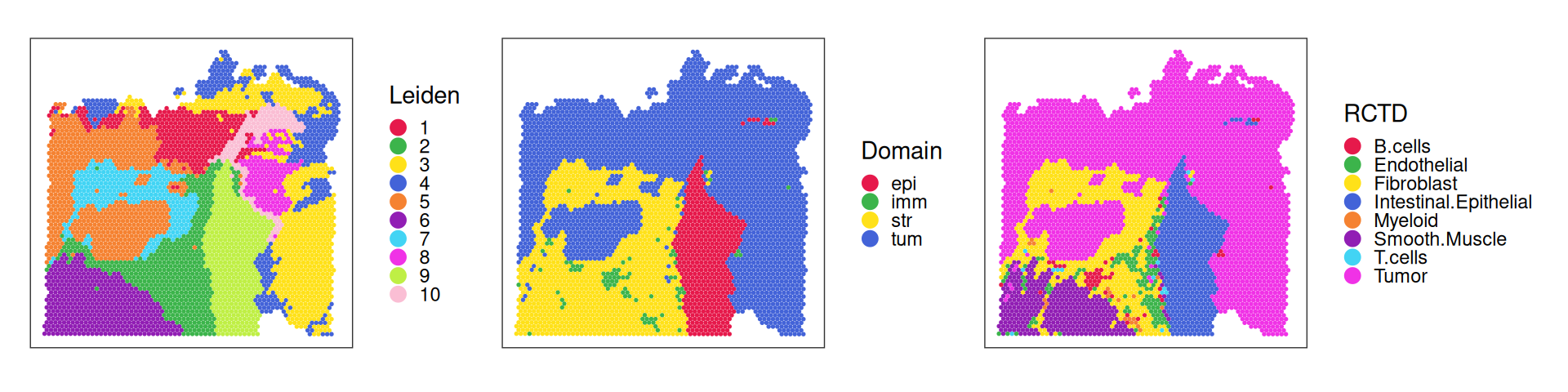
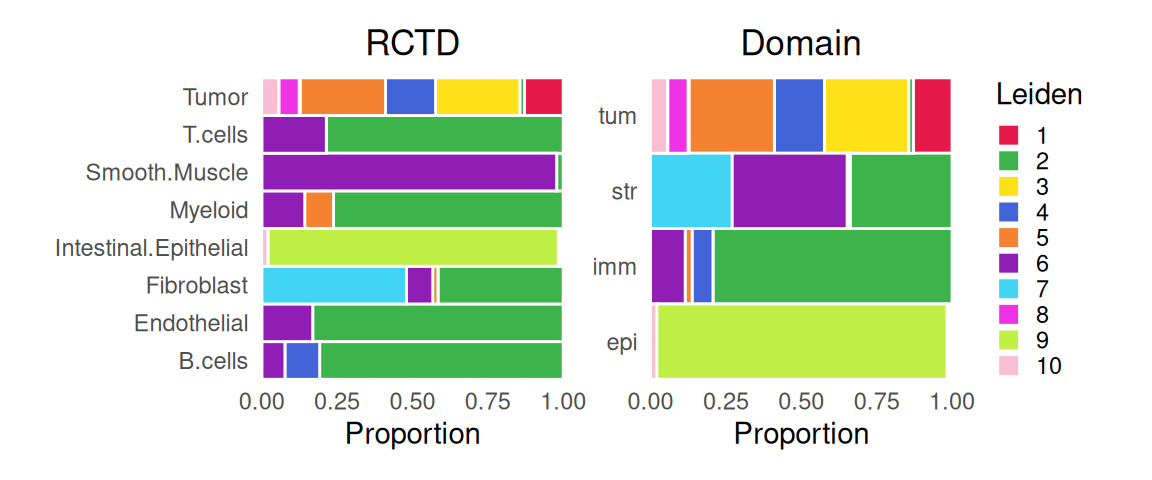
For comparison with unsupervised clustering (SNN-based Leiden), we also include assignments we would obtain if we were to assign spots the most frequent label (in terms of deconvolution estimates):
To help characterize subpopulations from unsupervised clustering, we can view their distribution across deconvolution-based clusters and broad domains; e.g., tumor spots are quite diverse, while smooth muscle spots and (normal) epithelia map almost completely to a single cluster:
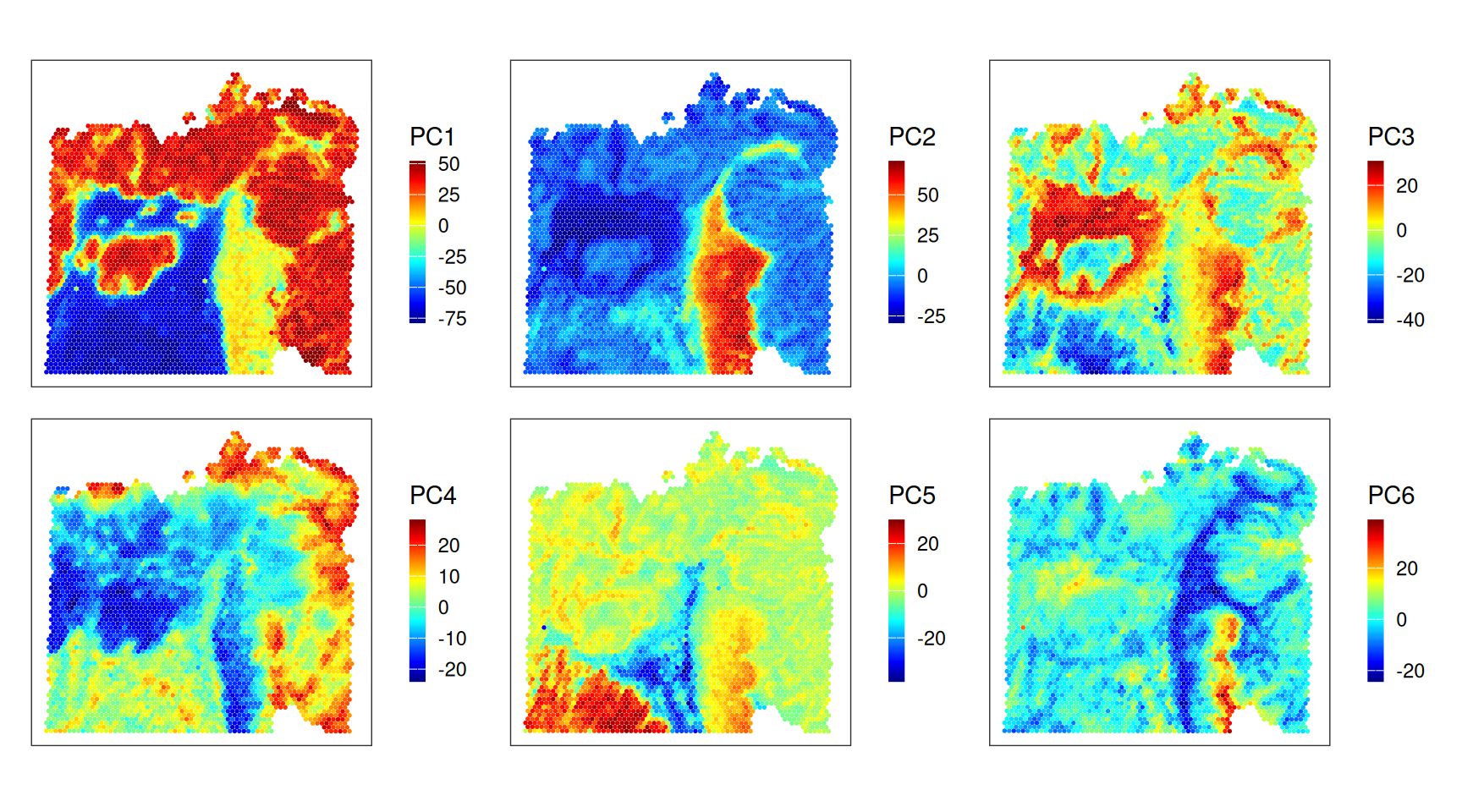
Let’s inspect the key drivers of (expression) variability in terms of PCs. Considering clustering and deconvolution results from above, we can see that
PC1 distinguishes stromal from both normal and malignant epithelia;
PC2 clearly separates (normal) intestinal epithelium from all else;
PC3 captures a fibroblast-rich region, and normal epithelia;
PC5 separates fibroblasts and smooth muscle cells; etc.
Quality control metrics tend to be low for specific clusters. Their patch-like pattern, in turn, explains the clustering of low-quality spots seen earlier.
Rather than investigating single genes, we can also evaluate the expression of sets of genes (e.g., pathway signatures); e.g., malignant tissue may differ in metabolic activity such as glycolysis and fatty acid metabolism, or exhibit increased apoptosis (cell death) etc. Here, we retrieve hallmark gene sets for some biological phenomena from MSigDB, using the msigdbr package:
Note that such gene lists like these may come from many different places - e.g., in-house analyses, other publications matching the research question etc. As such, they may also be read in from a .csv file, or stem from upstream analyses. In any case, they should be curated well and interpreted with caution.
Code
# retrieve hallmark gene sets from 'MSigDB'db<-msigdbr(species="Homo sapiens", collection="H")# get list of gene symbols, one element per setgs<-split(db$ensembl_gene, db$gs_name)# simplify set identifiers (drop prefix, use lower case)names(gs)<-tolower(gsub("HALLMARK_", "", names(gs)))# how many sets?length(gs)
## [1] 50
Code
# how many genes in each?range(sapply(gs, length))
## [1] 32 201
Next, we will score these using AUCell(Aibar et al. 2017), which works in two steps: (i) rank genes for every observation (here, spots), and (ii) compute AUC values for each gene set. In essence, these represent the fraction of genes (within top-ranked genes; default 5%) that are in a given set; i.e., high values correspond to high activity (in terms of coordinated gene expression).
By definition, AUCell yields values in [0,1]. Larger sets (more genes) are more likely to achieve higher scores by chance (e.g., a gene set of all genes would score 1 in any dataset). It is thus unfair to compare them directly. However, spatial distribution, correlation, and relative comparisons between subpopulations etc. are still meaningful.
Code
# realize (sparse) gene expression matrixmtx<-as(logcounts(spe), "dgCMatrix")# use ensembl identifiers as feature namesrownames(mtx)<-rowData(spe)$ID# build per-spot gene rankingsrnk<-AUCell_buildRankings(mtx, BPPARAM=bp, plotStats=FALSE, verbose=FALSE)# calculate AUC for each gene set in each spotauc<-AUCell_calcAUC(geneSets=gs, rankings=rnk, nCores=th, verbose=FALSE)# add results as spot metadatacolData(spe)[rownames(auc)]<-res<-t(assay(auc))
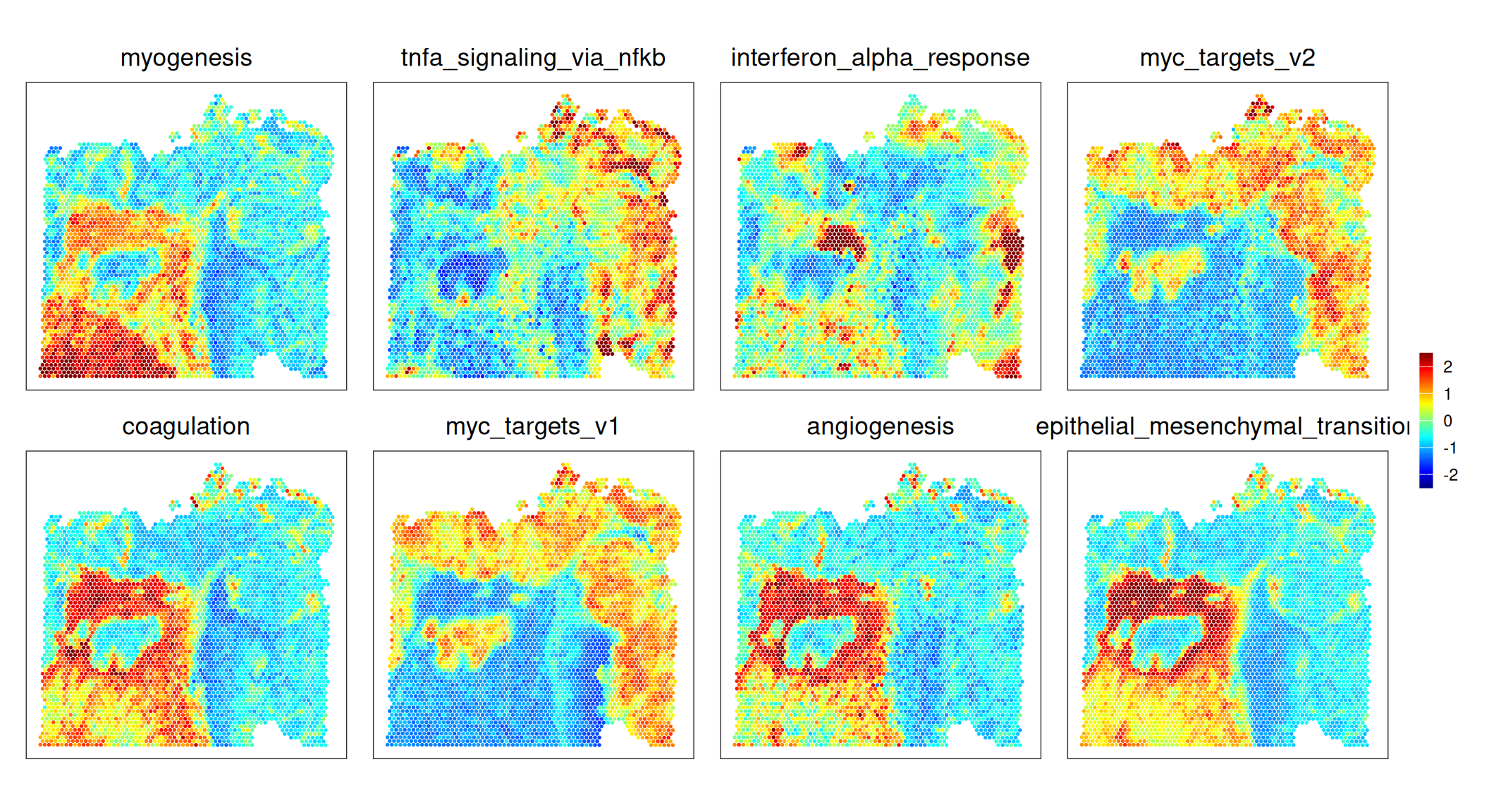
For simplicity, we’ll continue investigating only those signatures with the highest score variability across spots:
Code
var<-colVars(res)# variance across spotstop<-names(tail(sort(var), 8))# top sets
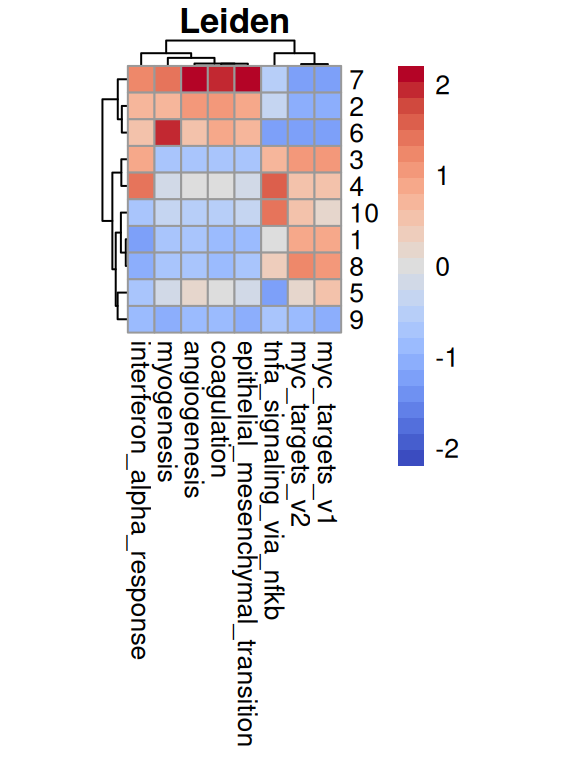
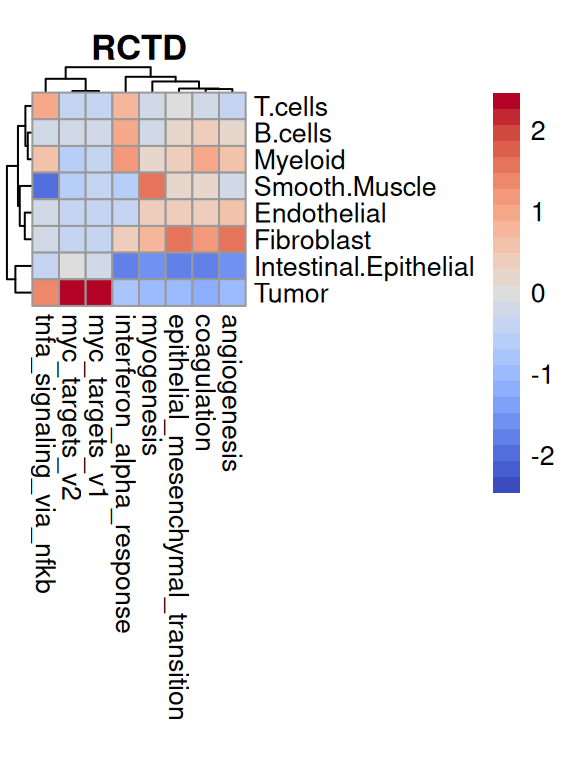
To summarize, MYC signaling is absent in stromal regions; the fibroblast ring surrounding a cancerous patch exhibits EMT, angiogenesis, etc.; INFa response and TNFa signaling is patch-like in both stroma and malignant epithelia.
To ease interpretability, we can stratify AUCell scores by spot labels; these may stem from an unsupervised or deconvolution-based approach:
Code
for(.inc("Leiden", "RCTD")){# aggregate AUC values by clusterks<-spe[[.]]pb<-aggregateAcrossCells.se(auc[top, ], ks, assay.type="AUC")mu<-sweep(assay(pb, "sums"), 2, pb$counts, `/`)colnames(mu)<-levels(ks)# visualize as (cluster x set) heatmappheatmap( mat=t(mu), scale="column", col=pals::coolwarm(), main=., cellwidth=10, cellheight=10, treeheight_row=5, treeheight_col=5)}
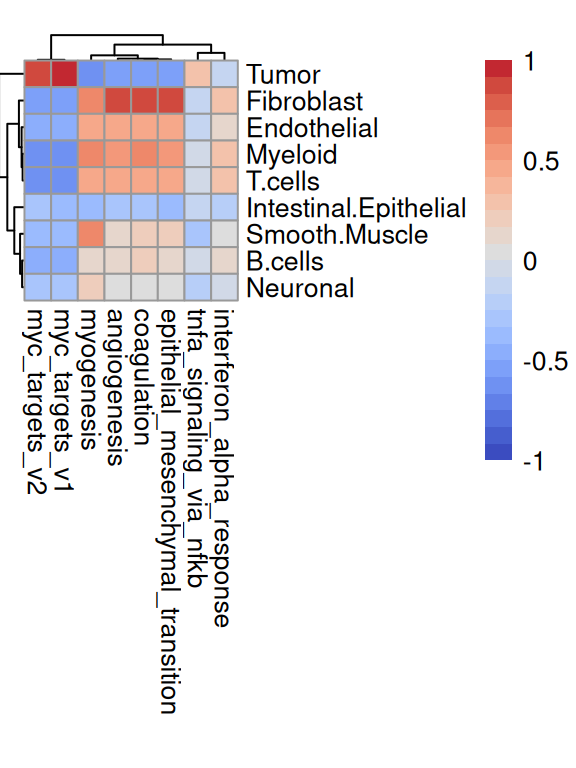
For the latter, we may instead correlate set scores with proportion estimates (rather than discretizing labels according to the dominant subpopulation):
Code
# correlate 'AUCell' signature scores with subpopulation# proportion estimates from deconvolution with 'RCTD'cm<-cor(as.matrix(ws), t(assay(auc[top, ])))
Aibar, Sara, Carmen Bravo González-Blas, Thomas Moerman, et al. 2017. “SCENIC: Single-Cell Regulatory Network Inference and Clustering.”Nature Methods 14: 1083–86. https://doi.org/10.1038/nmeth.4463.
Cable, Dylan M., Evan Murray, Luli S. Zou, et al. 2022. “Robust Decomposition of Cell Type Mixtures in Spatial Transcriptomics.”Nature Biotechnology 40: 517–26. https://doi.org/10.1038/s41587-021-00830-w.
de Oliveira, Michelli Faria, Juan Pablo Romero, Meii Chung, et al. 2025. “High-Definition Spatial Transcriptomic Profiling of Immune Cell Populations in Colorectal Cancer.”Nature Genetics 57: 1512–23. https://doi.org/10.1038/s41588-025-02193-3.