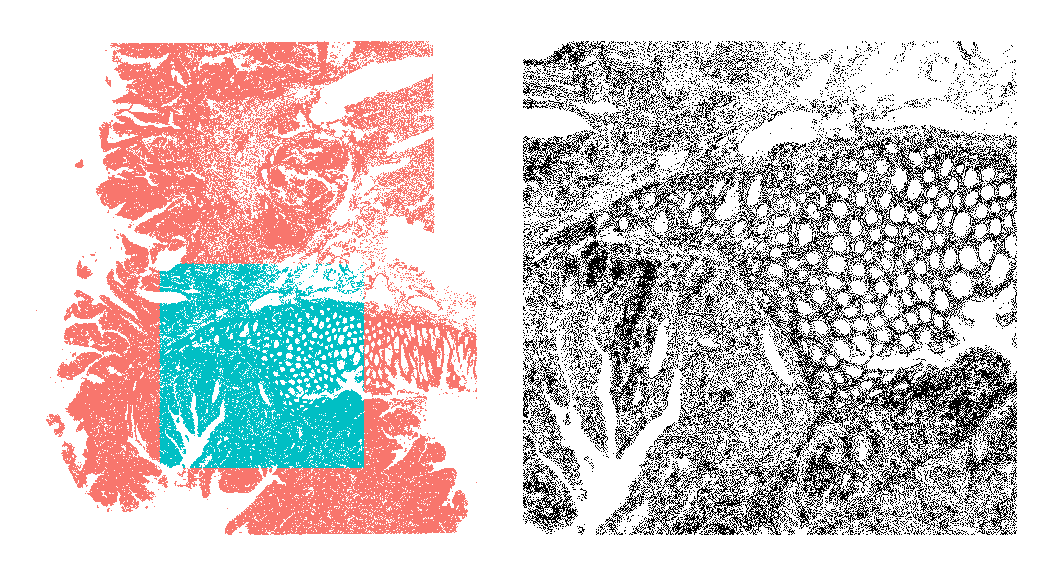In this demo, we will analyze a 313-plex Xenium dataset on human colorectal cancer tissue (Janesick et al. 2023). Following very basic quality control and preprocessing, we will perform both (non-spatial) unsupervised clustering as well as fully supervised label-transfer based on scRNA-seq reference data, and then compare the obtained cluster assignments to those provided by the authors.
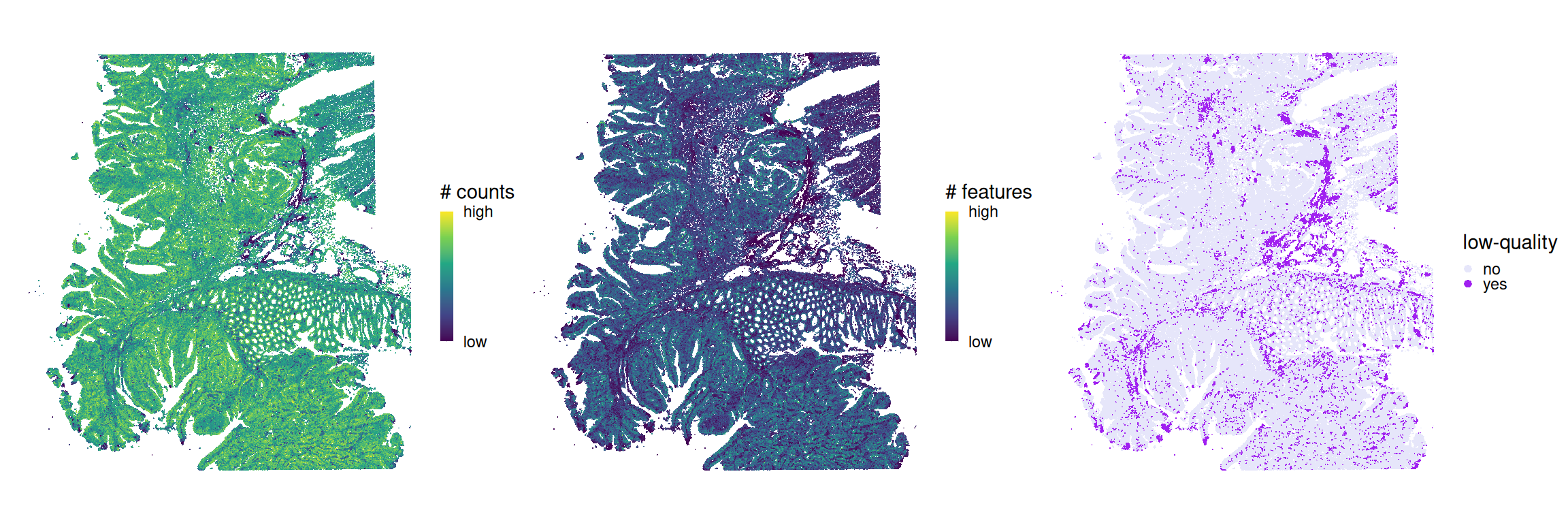
# compute cell-level QC metrics &# identify low-quality cells by thresholding on# median absolute deviation (MAD) from the medianspe<-quickRnaQc.se(spe, subsets=list())# tabulate # and % of cells discarded # due to few counts/detected featuresround(100*prop.table(table(keep=!spe$keep)), 2)
## keep
## FALSE TRUE
## 95.51 4.49
Before proceeding to exclude any cells from downstream analyses, let’s first visualize the cells deemed to be of high quality in space alongside the underlying quality control metrics (total counts and detected features):
Next, we’ll log-normalize counts by area, and perform principal component analysis (PCA) on all 422 RNA targets:
Code
# cell area-based normalizationsfs<-(.<-sub$cell_area)/median(.)sub<-normalizeRnaCounts.se(sub, size.factors=sfs)# principal component analysissub<-runPca.se(sub, features=rownames(sub))
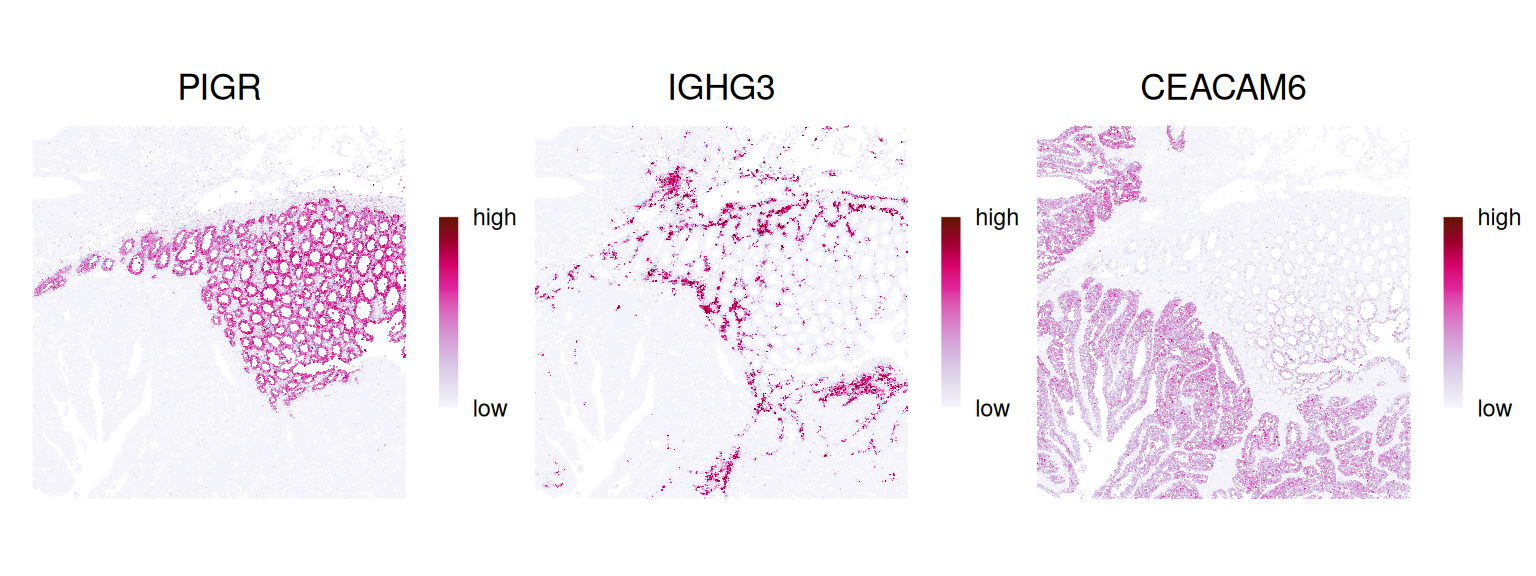
Let’s visualize the expression of some genes in space; e.g., PIGR, IGHG3 and CEACAM6, which should mark epithelial, plasma and tumor cells, respectively:
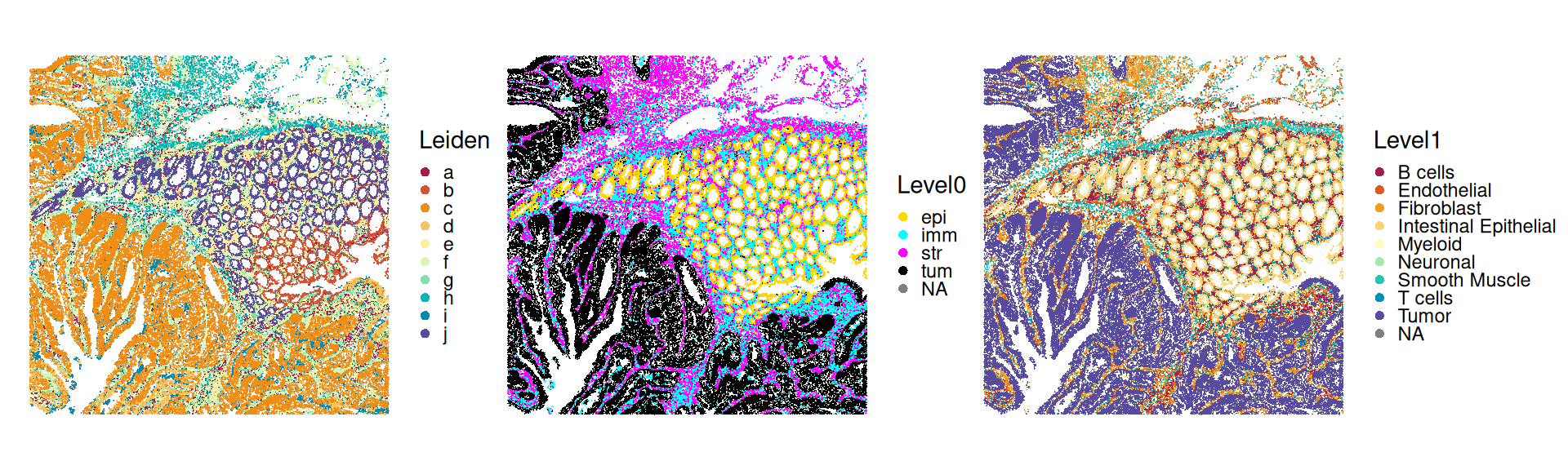
# shared nearest-neighbor (SNN) graph based on # cell-to-cell Jaccard similarity in PC space;# community detection using Leiden algorithmsub<-clusterGraph.se(sub, num.threads=th, resolution=0.5, method="leiden", output.name="Leiden", more.build.args=list(weight.scheme="jaccard"))table(sub$Leiden)
For comparison, we annotate the Xenium data using a label transfer approach, SingleR, which relies on labeled scRNA-seq data to compute references profiles and transfers labels based on the rank correlation between observed (here, Xenium) and reference (scRNA-seq) expression profiles.
First, we retrieve a matching (Chromium) scRNA-seq dataset, which includes low- (Level1) and high-resolution (Level2) annotations of cells into 9 and 31 subpopulations, respectively:
Here, we run SingleR using Level2 (high-resolution) annotations and with argument aggr.ref=TRUE, such that reference profiles will be aggregated (per cluster) prior to annotation. In this way, every Xenium cell will be assigned a label based on which pseudo-bulk scRNA-seq profile represents the best match.
Note that we filter the reference data to contain only cells from the same patient. This is not strictly necessary, assuming that clusters are transcriptionally stable across patients, but is done here to reduce runtime.
Code
# exclude cells deemed to be of low-qualitysce<-sce[, sce$QCFilter=="Keep"]# subset cells from same patientsce<-sce[, grepl("P2", sce$Patient)]# realize count matrixassay(sce)<-as(assay(sce), "dgCMatrix")# log-library size normalizationsce<-normalizeRnaCounts.se(sce)# restrict to Xenium targetssce<-sce[rowData(sce)$ID%in%rowData(sub)$ID, ]# set gene symbols as feature namesrownames(sce)<-rowData(sce)$Symbol# perform label transfer at the single cell-level,# using pseudo-bulk Chromium profiles as referenceres<-SingleR( test=sub, ref=sce, labels=sce$Level2, de.method="wilcox", aggr.ref=TRUE, BPPARAM=bp)
## Warning in scrapper::clusterKmeans(pcs, k = cur.ncenters, num.threads =
## num.threads): convergence failure for k-means
## Warning in scrapper::clusterKmeans(pcs, k = cur.ncenters, num.threads =
## num.threads): convergence failure for k-means
## Warning in scrapper::clusterKmeans(pcs, k = cur.ncenters, num.threads =
## num.threads): convergence failure for k-means
## Warning in scrapper::clusterKmeans(pcs, k = cur.ncenters, num.threads =
## num.threads): convergence failure for k-means
Simplifying further, we can group cells into different compartments, namely, (malignant) tumor, immune, epithelial and stromal cells; we’ll see below that visualizing cells in this way nicely captures the general tissue structure.
Tabulating the cluster assignments between Leiden (unsupervised) and SingleR (supervised), we can observe overall high concordance; i.e., most clusters have a one-to-one mapping between both approaches. However, some subpopulations are split between clusters; e.g., cells labeled as cluster fibroblasts, endothelia and smooth muscle cells by SingleR tend to intermix in the Leiden clusters. This is not unexpected, given that these are all stromal subpopulations with comparatively similar transcriptional profiles. (Note that we are observing a mere fraction of the whole transcriptome with the Xenium panel employed here.)
Code
# contingency table & number of clustersround(100*prop.table(table(sub$Level1, sub$Leiden), 2), 1)
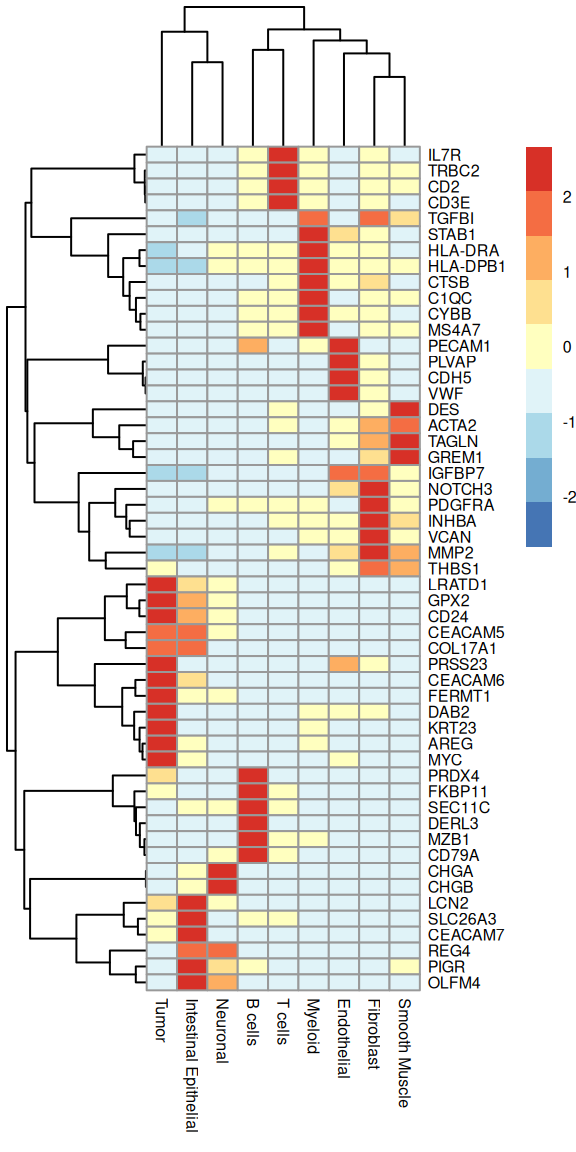
Below, we test for differential expression between Level1 clusters, and visualize selected markers as a heatmap of (z-scaled) average expression. It’s comforting to see that we pick up on many classics, e.g., endothelia are marked by VWF and PECAM1, T cells by CD2 and TRAC, etc.
Code
# test for differential expression between clustersok<-!is.na(sub$Level1)de<-scoreMarkers.se(sub[, ok], sub$Level1[ok])# select top-ranked genes for every clustergs<-lapply(de, \(df)head(rownames(df), 5))gs<-unique(unlist(gs))# visualize their average expression by clusterplotGroupedHeatmap(sub, features=gs, group="Level1", scale=TRUE, center=TRUE, fontsize=6)
25.6 Appendix
References
Janesick, Amanda, Robert Shelansky, Andrew D. Gottscho, et al. 2023. “High Resolution Mapping of the Tumor Microenvironment Using Integrated Single-Cell, Spatial and in Situ Analysis.”Nature Communications 14 (8353). https://doi.org/10.1038/s41467-023-43458-x.