37 Structure-based analysis
37.1 Preamble
37.1.1 Introduction
Spatial omics data allow us to quantify various features that are related to tissue architecture. This allows us to perform comparisons of anatomical structure-derived features. We will refer to this type of analysis as structure-based analysis.
We will use the sosta package (Gunz et al. 2025) to reconstruct and quantify pancreatic islets from human donors with different stages of type 1 diabetes (T1D) and healthy controls (Damond et al. 2019). We will then compare some of the features between conditions.
This chapter is closely related to the spatial statistics and neighborhood concepts introduced in Chapter 32 and Chapter 22, but it summarizes those spatial patterns as structure-level geometric features before comparing groups of samples.
37.1.2 Dependencies
37.1.3 Load data
An assay-free version of the dataset is available through our OSF repository (used here to reduce runtime as the following analyses do not require assay data):
Code
# load data from OSF repository (for smaller download)
osf_repo <- osf_retrieve_node("https://osf.io/5n4q3/")
osf_files <- osf_ls_files(osf_repo,
path="zzz", n_max=Inf,
pattern="Damond_noAssays.rds")
dir.create(td <- tempfile())
foo <- osf_download(osf_files, path=td)
spe <- readRDS(file.path(td, list.files(td, ".rds")))
table(spe$patient_id, spe$patient_stage)##
## Long-duration Non-diabetic Onset
## 6089 91634 0 0
## 6126 0 158693 0
## 6134 0 181609 0
## 6180 141450 0 0
## 6228 0 0 142106
## 6264 146123 0 0
## 6278 0 185399 0
## 6362 0 0 202272
## 6380 0 0 129456
## 6386 0 127350 0
## 6414 0 0 166052
## 6418 104830 0 037.2 Visualize data
First, we plot a few selected fields of view (FOVs).
Code
# subset to 4 randomly selected FOVs
fov <- sample(unique(spe$image_name), 4)
sub <- spe[, spe$image_name %in% fov]
df <- data.frame(colData(sub), spatialCoords(sub))
# visualize annotations
ggplot(df, aes(cell_x, cell_y, color=cell_category)) +
geom_point(size=0.4) + facet_wrap(~image_name, ncol=2) +
scale_color_manual(values=unname(pals::okabe(n=5))) +
guides(col=guide_legend(override.aes=list(size=2))) +
coord_equal() + theme_classic() 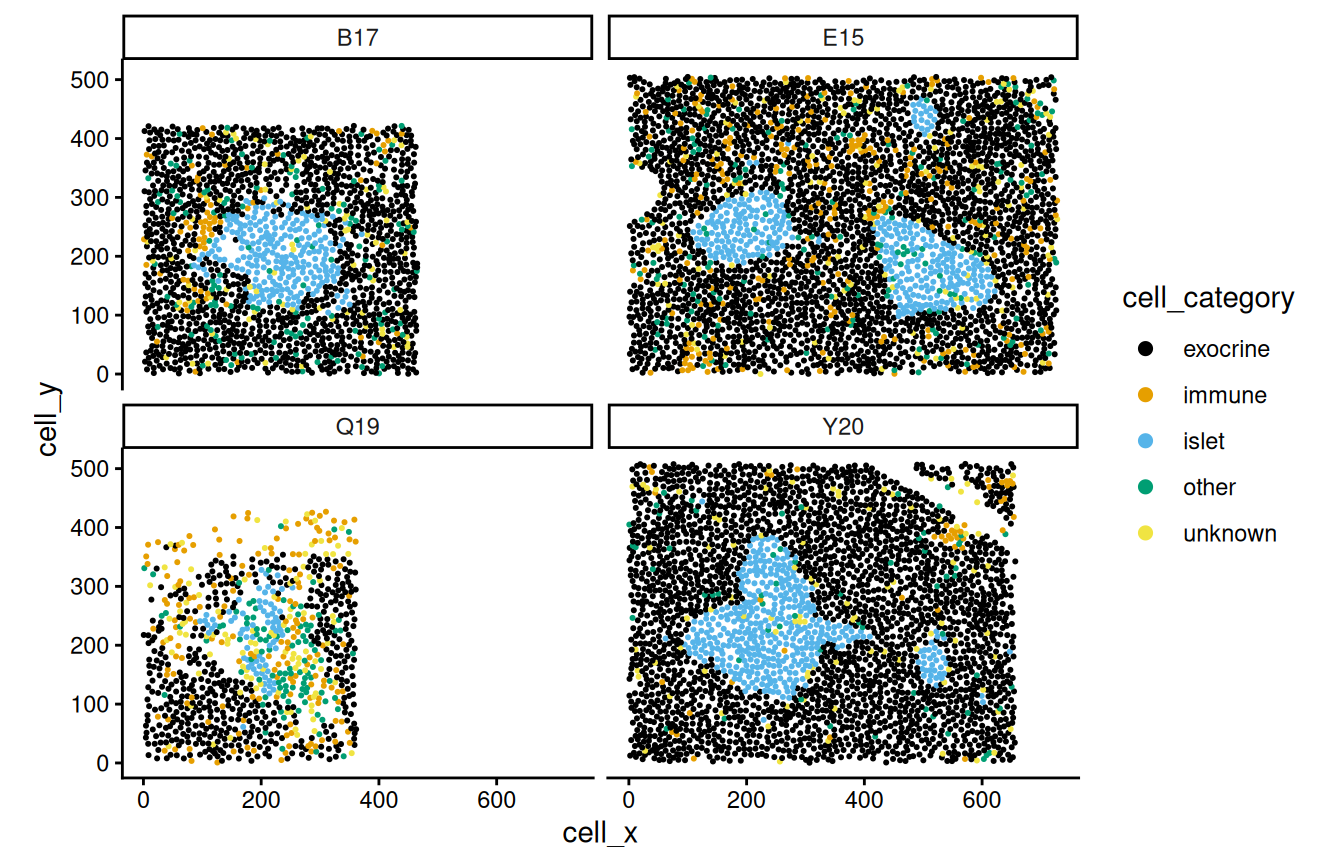
37.3 Islet reconstruction
Next, we will reconstruct or segment the individual islets using a density-based approach implemented in sosta.
Code
# randomly sample 'nImages' images
# for parameter estimation
est <- estimateReconstructionParametersSPE(
spe,
marks="cell_category",
imageCol="image_name",
markSelect="islet",
nImages=10,
nCores=4,
plotHist=FALSE)
# get parameter estimates for ...
th <- mean(est$thres) # threshold
bw <- mean(est$bndw) # bandwidthThe sosta package uses a reconstruction method based on the point pattern density of the islet cells. This method needs two parameters:
- a bandwidth that is used for intensity profile estimation, and
- a threshold based on which the islets are reconstructed.
Code
shapeIntensityImage(
spe,
marks="cell_category",
imageCol="image_name",
imageId=fov[4],
markSelect="islet")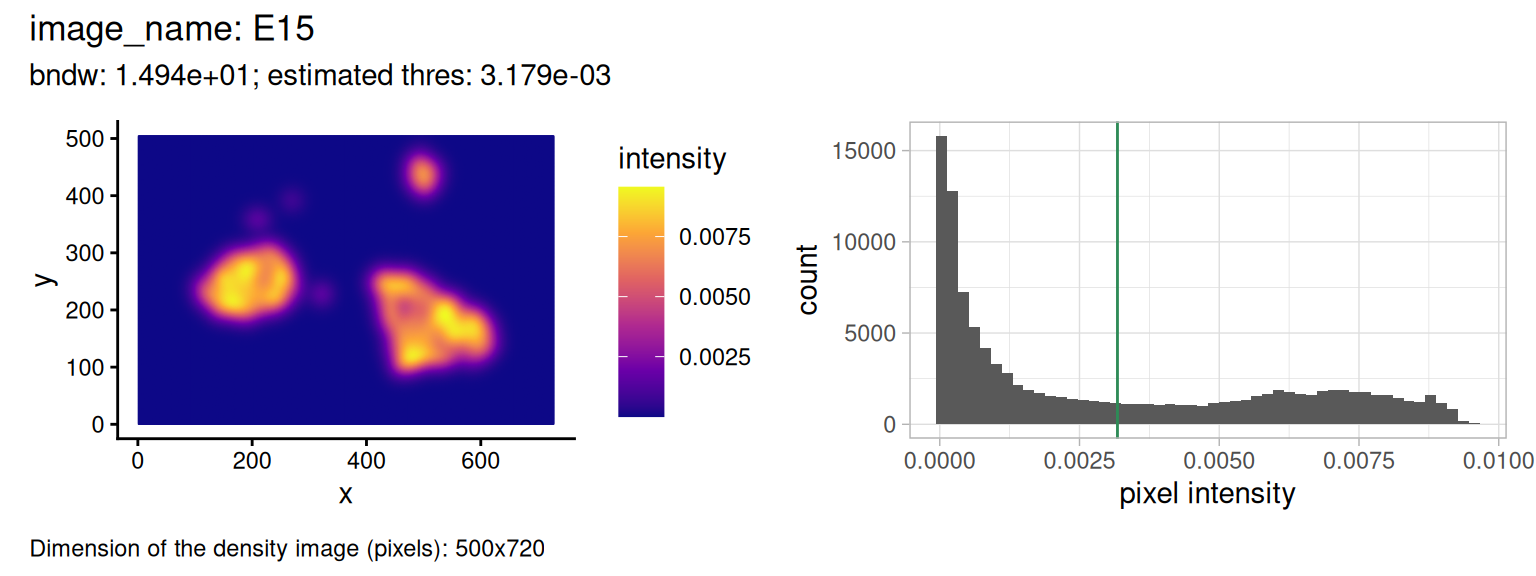
Shown on the left is a density (pixel-level) image, and on the right a histogram of the intensity values. The method selects every pixel above a certain threshold for reconstruction. The smoothing bandwidth that generates the image is estimated using cross-validation (see function bw.diggle() of the spatstat.explore package from CRAN).
The threshold is estimated by taking the mean between the two modes of the (truncated) pixel intensity distribution. The function estimateReconstructionParametersSPE() repeats the estimation of these parameters for a selection of images in the dataset.
To make the reconstruction comparable between images we then use a fixed set of parameters for all images, in our case the mean of the individual parameters for a subset of the dataset.
Code
allIslets <- reconstructShapeDensitySPE(
spe,
marks="cell_category",
imageCol="image_name",
markSelect="islet",
bndw=bw,
thres=th,
nCores=4)Now we can inspect the reconstruction in the sample images.
Code
# subset to selected FOVs
subIslets <- allIslets[allIslets$image_name %in% fov, ]
# visualize annotations
ggplot(df, aes(cell_x, cell_y, color=cell_category)) +
geom_point(size=0.4) + facet_wrap(~image_name, ncol=2) +
scale_color_manual(values=unname(pals::okabe(n=5))) +
guides(col=guide_legend(override.aes=list(size=2))) +
coord_equal() + theme_classic() +
geom_sf( # geom for structure outlines
data=subIslets, inherit.aes=FALSE,
color="red", fill=NA, linewidth=1)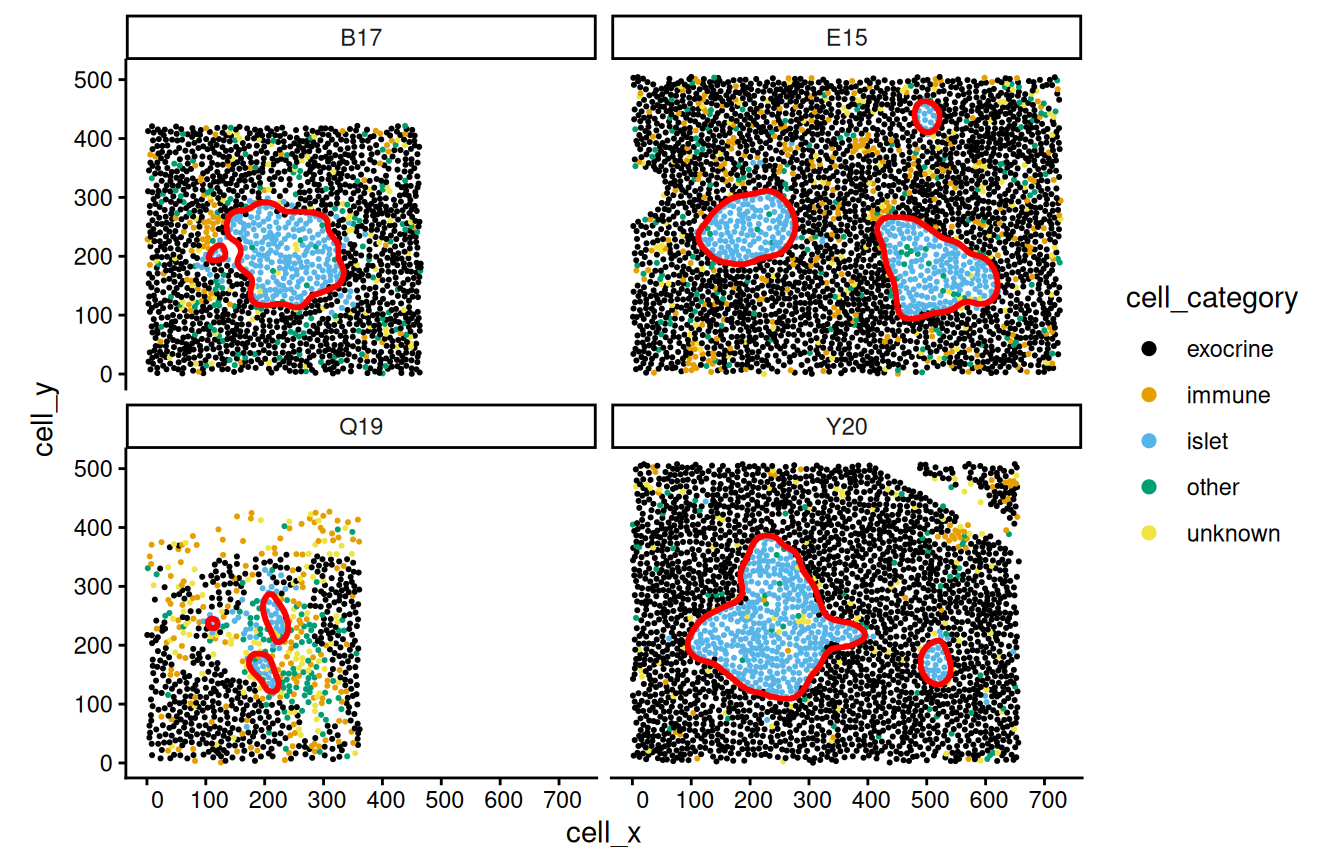
The allIslets object is a simple feature collection which contains polygons (<GEOMETRY> column), a structure identifier (structID), and the image identifier (image_name). We will add some patient metadata to the object.
Code
# factor levels for 'patient_stage'
lv <- c("Non-diabetic", "Onset", "Long-duration")
# cell metadata columns to keep
colsKeep <- c(
"sample_id", "image_name",
"patient_disease_duration",
"patient_id", "patient_stage",
"patient_age", "patient_gender",
"tissue_slide", "tissue_region")
patientData <- colData(spe) |>
as_tibble() |>
# keep selected columns
select(all_of(colsKeep)) |>
# refactor patient IDs & stages
group_by(image_name) |>
mutate_at("patient_id", factor) |>
mutate_at("patient_stage", factor, lv) |>
# keep only unique combinations
unique()
# join with results from reconstruction
allIslets <- left_join(allIslets, patientData, by="image_name")37.4 Quantification of geometric features
Now we can proceed with quantification of geometric aspects of the islets, and combine them with patient information.
Code
isletMetrics <- totalShapeMetrics(allIslets)
allIslets <- cbind(allIslets, t(isletMetrics))PCA can give us an overview of the different features; here, each dot represents one structure.
Code
pca <- prcomp(t(isletMetrics), scale.=TRUE)
autoplot(pca,
x=1, y=2,
data=allIslets,
color="patient_stage",
size=2,
loadings=TRUE,
loadings.colour="steelblue3",
loadings.label=TRUE,
loadings.label.size=3,
loadings.label.repel=TRUE,
loadings.label.colour="black") +
theme_bw() + coord_fixed() +
scale_color_manual(values=unname(pals::tol(n=3)))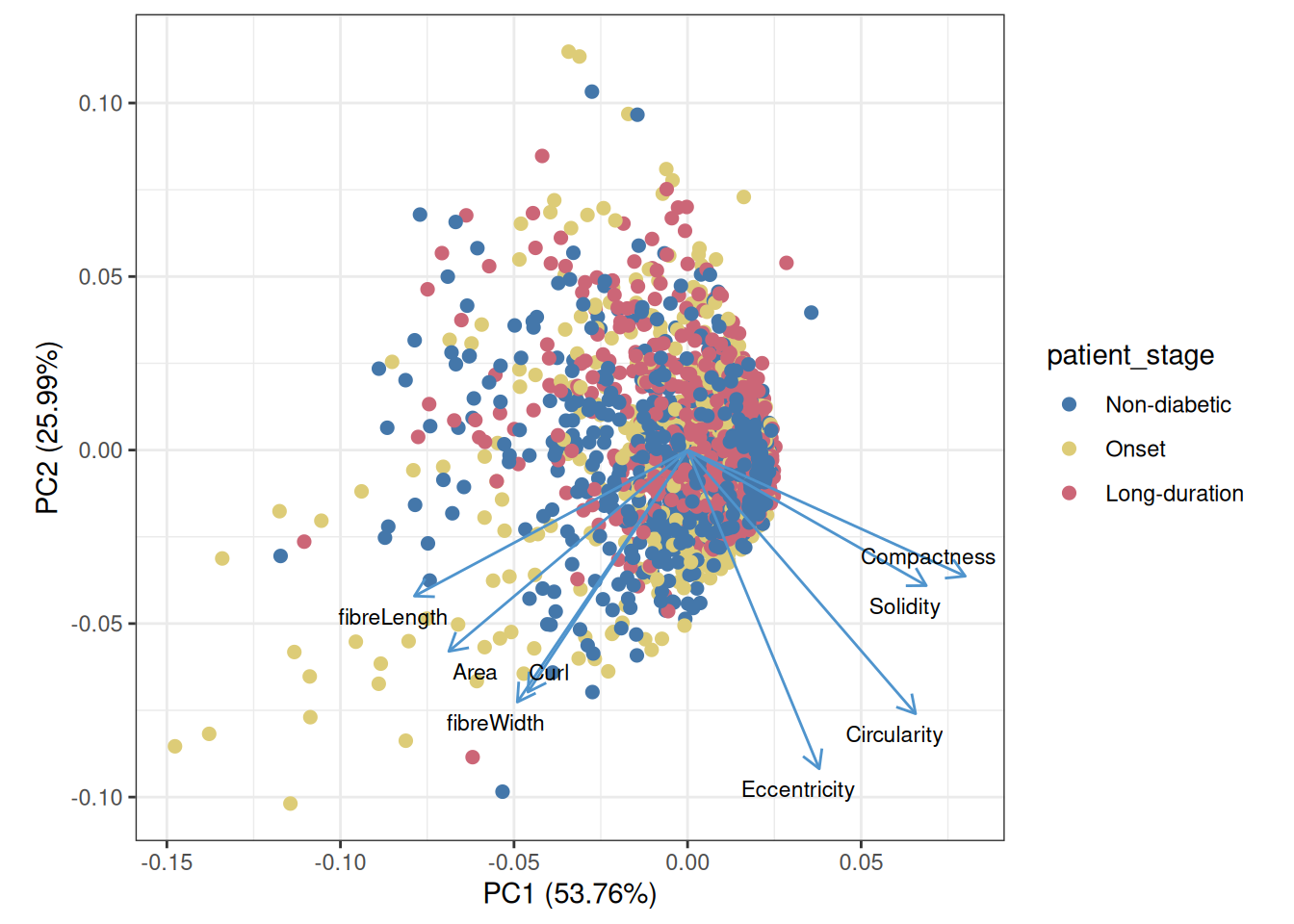
Code
# wrangling
df <- allIslets |>
st_drop_geometry() |>
select(patient_stage, rownames(isletMetrics)) |>
pivot_longer(-patient_stage) |>
filter(name %in% c("Area", "Compactness", "Curl"))
# visualization
ggplot(df, aes(patient_stage, value, fill=patient_stage)) +
geom_violin() + geom_boxplot(aes(fill=NULL), width=0.3) +
scale_fill_manual(values=unname(pals::tol(n=3))) +
scale_x_discrete(guide=guide_axis(n.dodge=2)) +
facet_wrap(~name, scales="free") +
guides(fill="none") + theme_bw()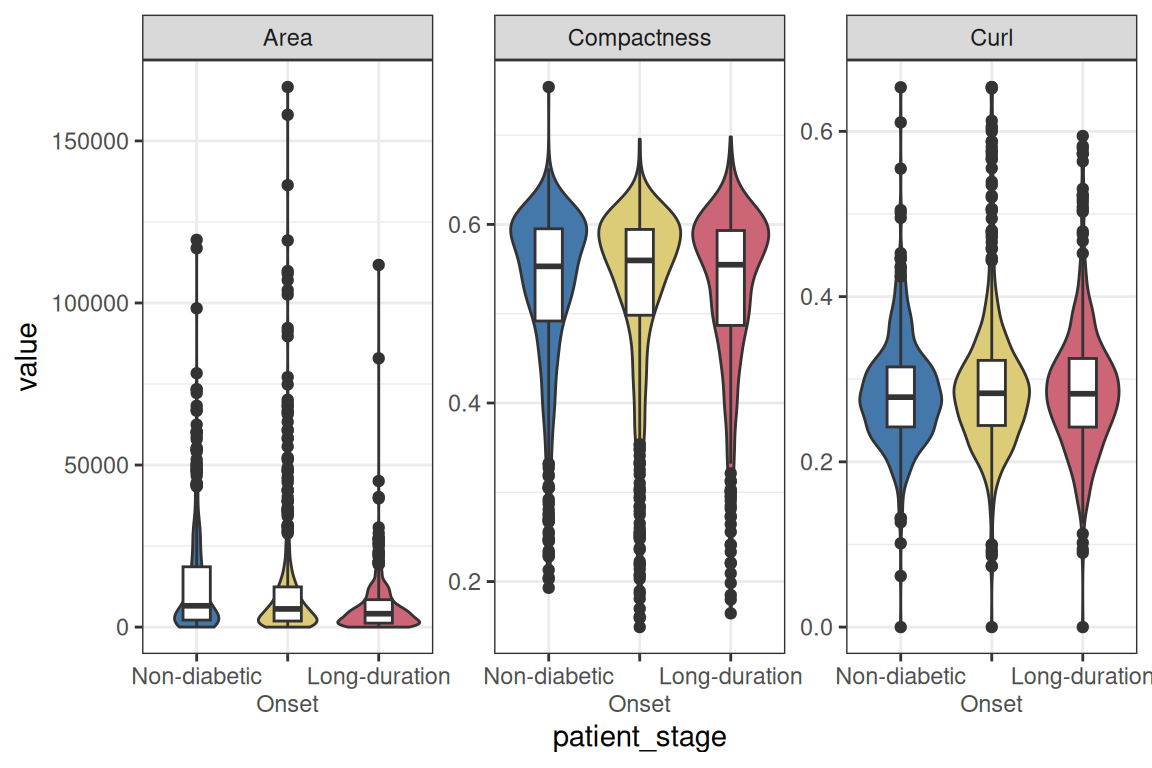
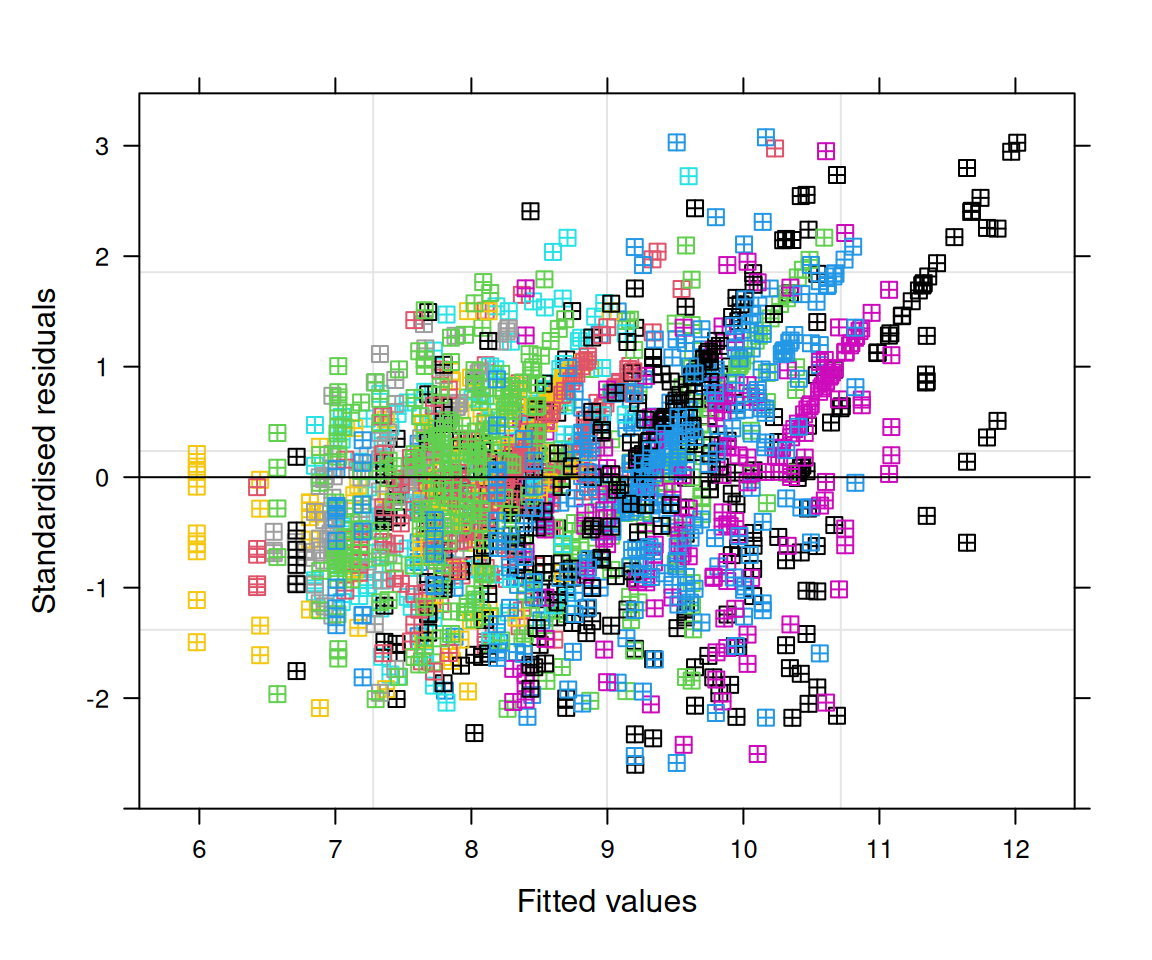
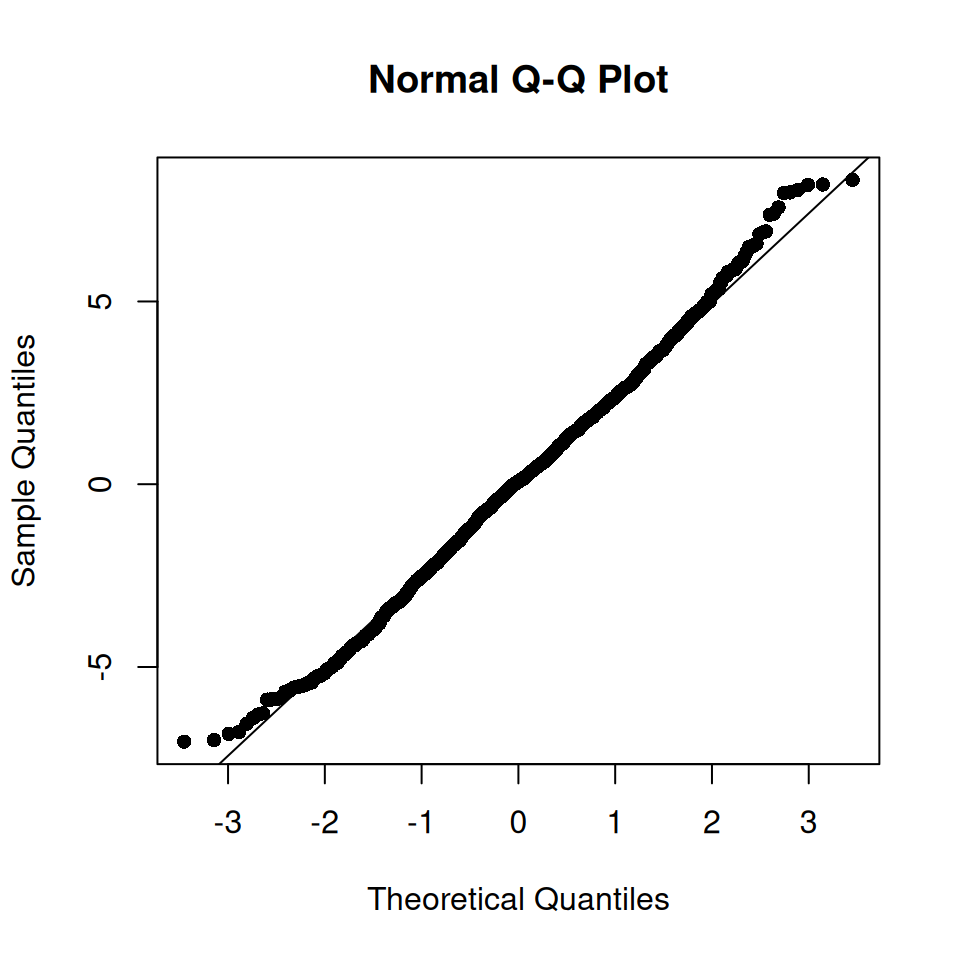
Let’s focus on the area of the islets and facet by stage to look at patient variability. As the distribution is very skewed, we will use a 1/4-power transformation on the area of the islets. The transformation was chosen after inspection of the model diagnostics (see below).
Code
# wrangling
df <- allIslets |>
st_drop_geometry() |>
select(patient_stage, patient_id, rownames(isletMetrics)) |>
pivot_longer(-c(patient_stage, patient_id)) |>
filter(name %in% c("Area"))
# visualization
ggplot(df, aes(patient_id, value^(1/4), fill=patient_stage)) +
geom_violin() + geom_boxplot(fill=NA, width=0.3) +
scale_fill_manual(values=unname(pals::tol(n=3))) +
facet_wrap(~patient_stage, scales="free_x") +
geom_jitter(size=0.5) + ylab("Area") +
guides(fill="none") + theme_bw()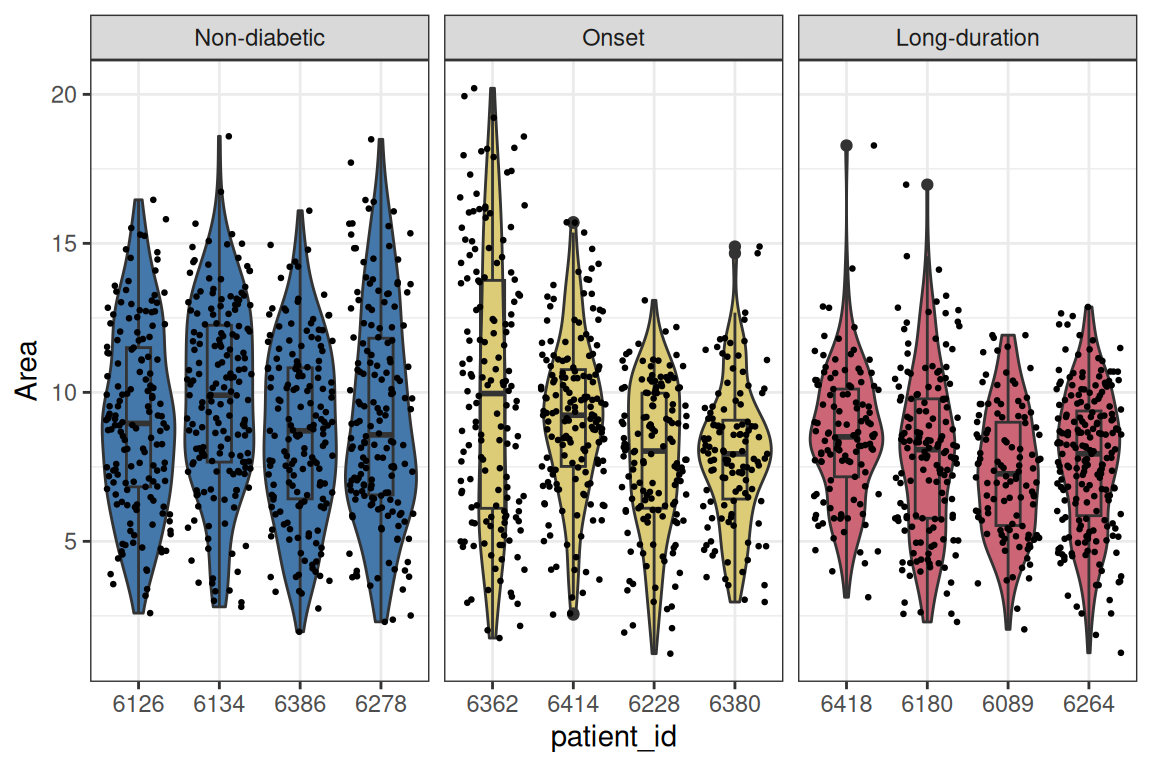
37.5 Between-sample comparison
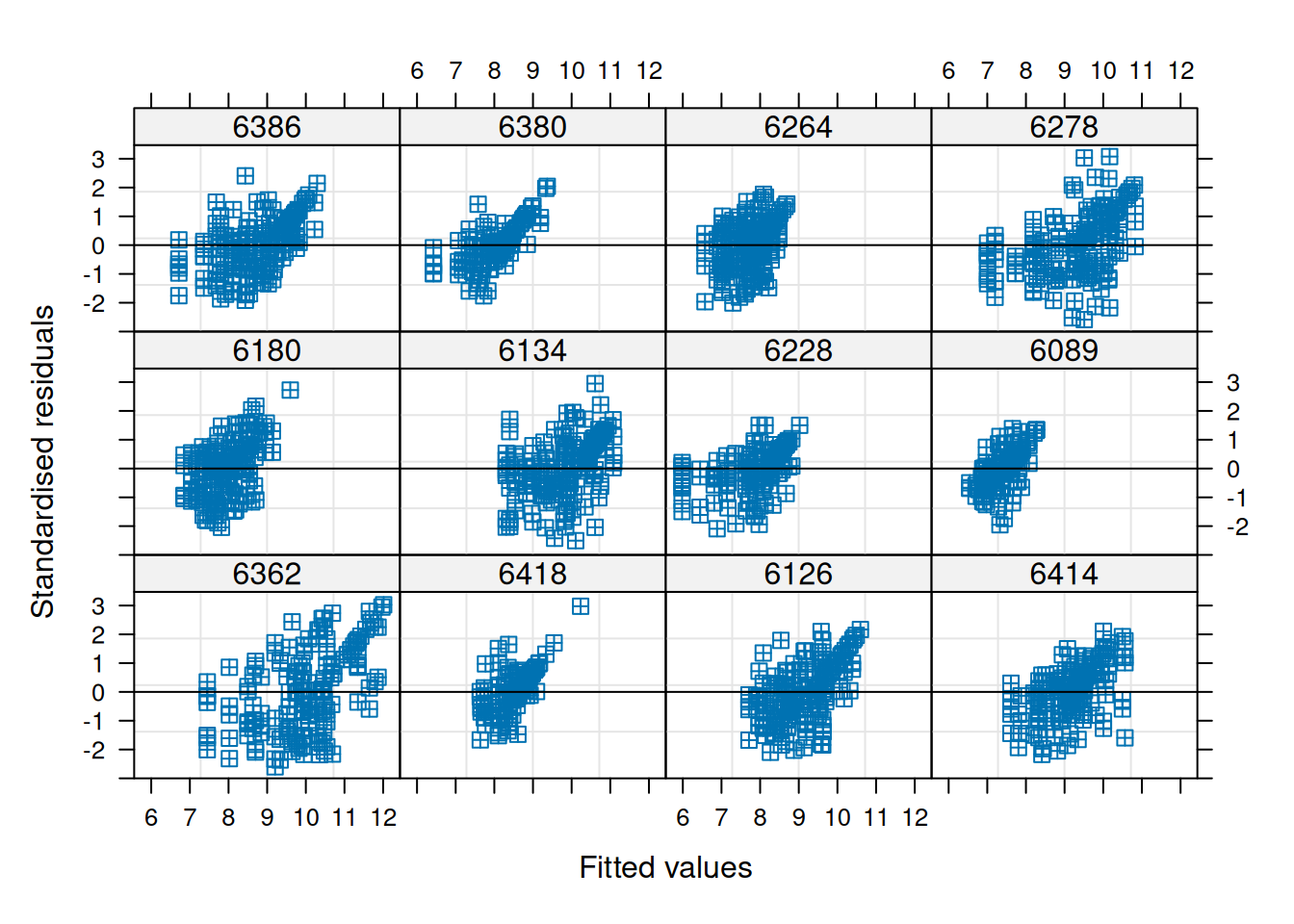
An important aspect to note is that the individual structure metrics are not independent measurements, since there are generally repeated measurements (i.e., multiple islets) per slide and per patient. Therefore, we need to account for this correlation between measurements.
To account for this, we will use mixed linear models with random effects for the patient and the individual slides (image name). The lme4 package will be used for fitting linear mixed effects models (Bates et al. 2015) and lmerTest for p-value calculations (Kuznetsova et al. 2017).
Code
## Linear mixed model fit by REML. t-tests use Satterthwaite's method [
## lmerModLmerTest]
## Formula: f
## Data: allIslets
##
## REML criterion at convergence: 9028
##
## Scaled residuals:
## Min 1Q Median 3Q Max
## -2.6027 -0.6199 0.0265 0.6129 3.0764
##
## Random effects:
## Groups Name Variance Std.Dev.
## image_name (Intercept) 1.4628 1.2095
## patient_id (Intercept) 0.5426 0.7366
## Residual 7.3230 2.7061
## Number of obs: 1806, groups: image_name, 844; patient_id, 12
##
## Fixed effects:
## Estimate Std. Error df t value Pr(>|t|)
## (Intercept) 9.5935 0.3924 8.8393 24.447 2.01e-09 ***
## patient_stageOnset -0.5624 0.5557 8.8860 -1.012 0.338
## patient_stageLong-duration -1.5404 0.5563 8.9198 -2.769 0.022 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Correlation of Fixed Effects:
## (Intr) ptnt_O
## ptnt_stgOns -0.706
## ptnt_stgLn- -0.705 0.498From the fixed effects section in the model summary above, we note that there is a statistically significant difference in the transformed islet area of long-duration patients compared to non-diabetic patients, while the effect for onset patients is not significant at the 5% level. This result accounts for correlation at the patient and image level as modeled by random intercepts. The transformation (1/4-power) was chosen after inspection of the residual behavior in the model diagnostics for this dataset and feature of interest specifically.