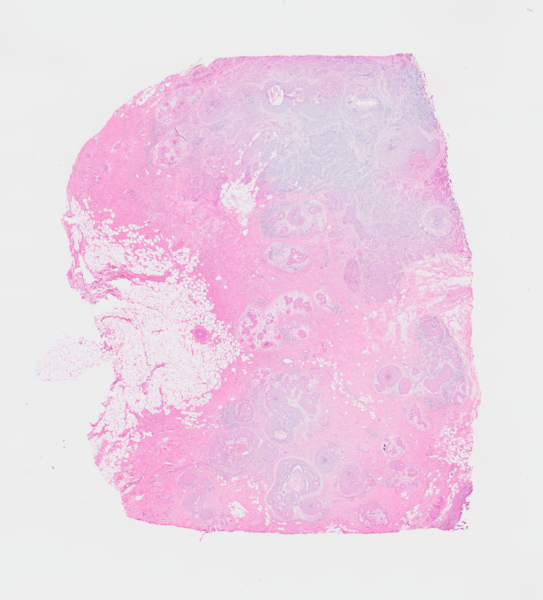
In this demo, we will rely on data from Janesick et al. (2023), which includes same-section Visium and Xenium measurements on human breast cancer tissue.
# also retrieve cell subpopulation labelsdf<-read.csv(file.path(td, "annotation.csv"))xen$anno<-df$Annotation[match(xen$cell_id, df$Barcode)]
We’ll also do some data wrangling to simplify spatial coordinate names, and use gene symbols (rather than ensembl identifiers) as feature names for both data:
To align Xenium and Visium sections, we use the affine transformation matrix provided by 10x Genomics, which was obtained by registration of Xenium onto Visium in Python with the Fiji Java plug-in; see Chapter 38 for details.
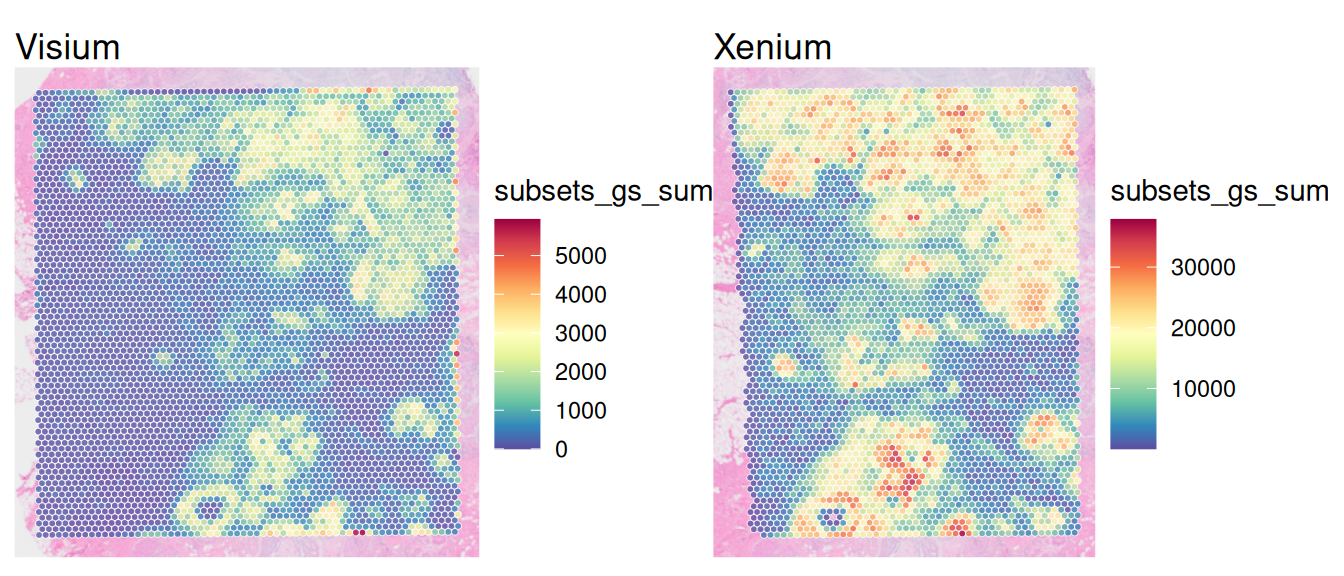
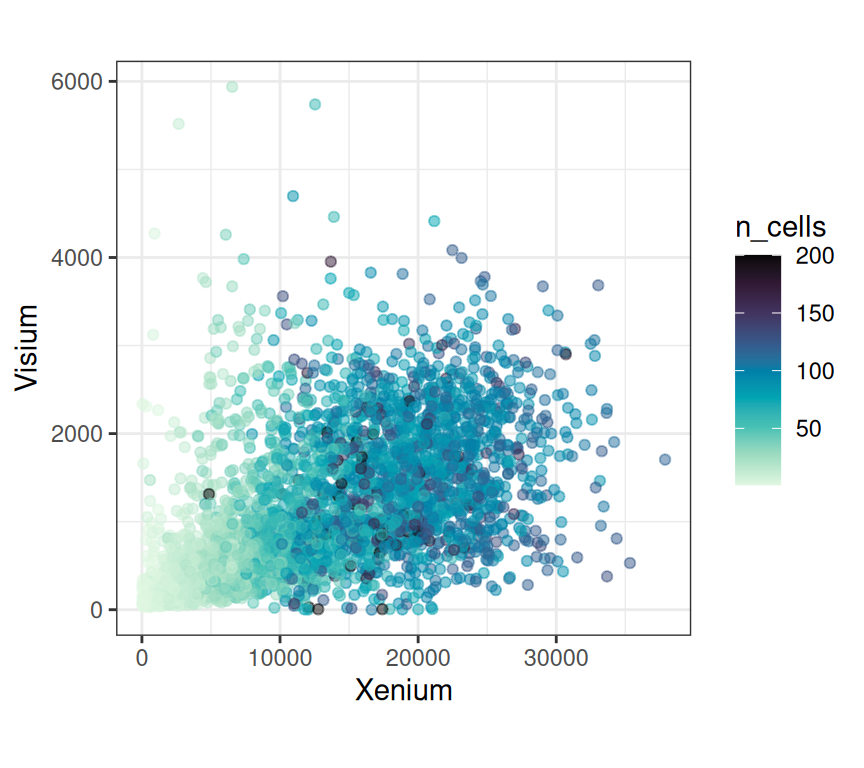
Binning single-cell resolution spatial data into spots can be useful for checking correlations between technical replicates of the same technology, identifying artifacts across technologies, and checking cell density (number of cells per spot). In general, it is possible to bin at the transcript-level (subcellular) or cell-level.
To aggregate single cell-level data from Xenium at the spot level, we first carry out a fixed-radius neighborhood search using RANN (see Chapter 22) to identify, for every spot, cells whose centroid lies within a \(\sim130\)um distance (Visium spot diameter of 55um, divided by two 2, divided by 0.2115 = Xenium px size in um):
Code
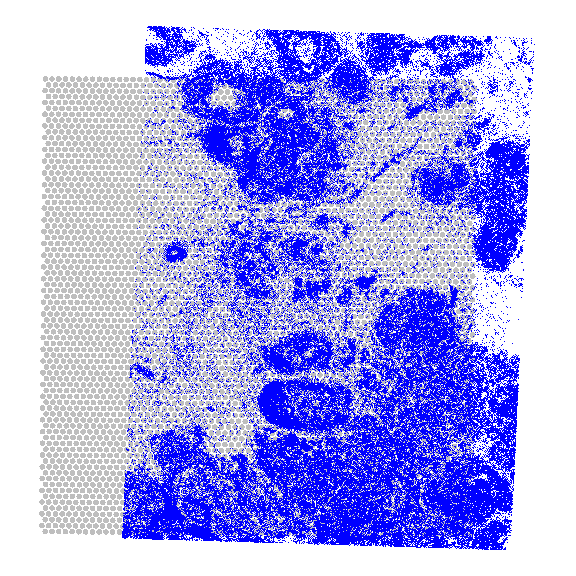
# do a fixed-radius search to get cell # centroids that fall on a given spotnns<-nn2( searchtype="radius", radius=55/2/0.2125, k=200, data=spatialCoords(xen), query=spatialCoords(vis))
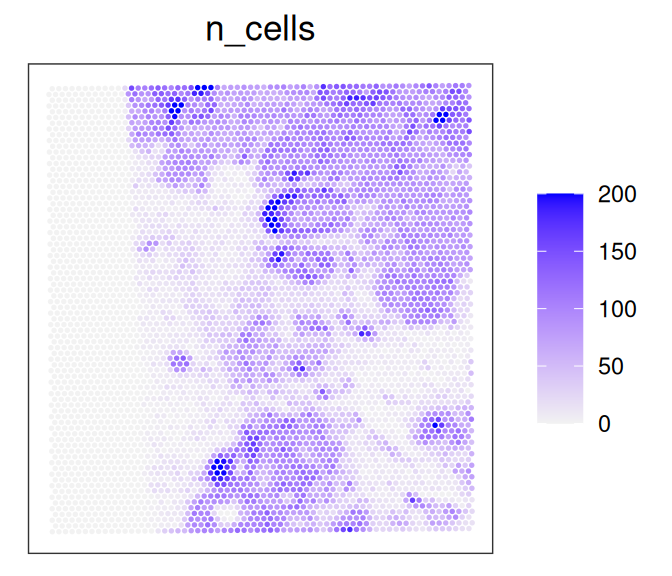
Let’s count and visualize the number of cells that overlap each spot:
Code
# get cell indices and number of cells per spotvis$n_cells<-rowSums((idx<-nns$nn.idx)>0)plotCoords(vis, annotate="n_cells")
Next, we can aggregate single cell-level Xenium data into pseudo-spots. In addition, we propagate the Visium data’s spatial coordinates, excluding spots without any overlapping cells:
Code
# aggregate Xenium data into pseudo-spotsids<-rep.int(seq(ncol(vis)), vis$n_cells)xem<-aggregateAcrossCells.se(xen[, c(t(idx))], ids)xem<-as(as(xem, "SingleCellExperiment"), "SpatialExperiment")# propagate Visium data's spatial coordinates, excluding empty pseudo-spotsspatialCoords(xem)<-spatialCoords(vis)[vis$n_cells>0, ]colnames(xem)<-colnames(vis)[vis$n_cells>0]assays(xem)<-list(counts=assay(xem))xem$in_tissue<-1
Because we’ve aligned the Xenium to the Visium data, we can also propagate the Visium data’s imgData (low resolution H&E staining) to the object containing pseudo-spot Xenium data:
Next, let’s compute some standard quality control metrics on both, the Visium and pseudo-spot Xenium data. Besides dataset-specific metrics, we also specify the subset of genes that are shared between both datasets in order to obtain comparable metrics:
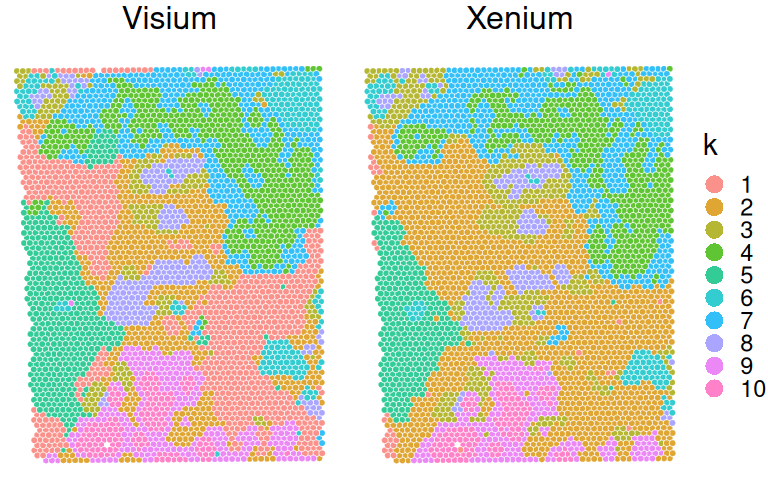
In order to perform joint spatial clustering of both modalities, we construct array coordinates for Xenium pseudo-spots that are offset from Visium spots:
Code
# offset the spatial location for joint clustering# of Visium and adjacent pseudo-spot Xenium dataobj$array_row<-c(ar<-vis[, cs]$array_row, 100+ar)obj$array_col<-c(ac<-vis[, cs]$array_col, 100+ac)
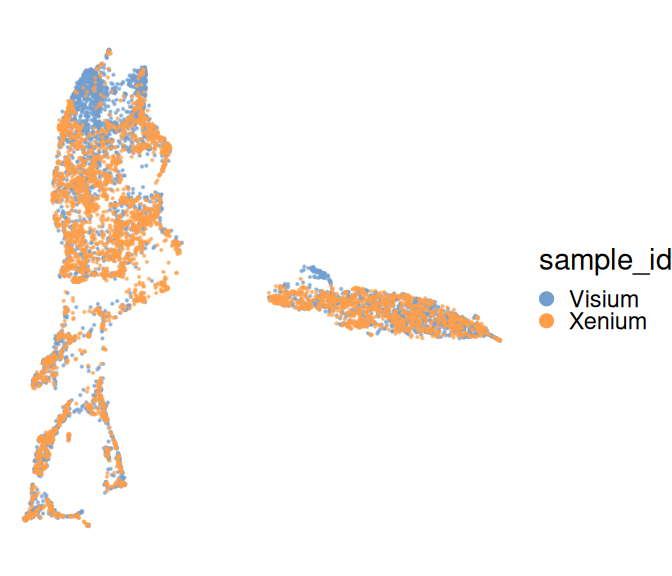
Next, we run a standard pipeline to perform log-library size normalization (using scrapper), principal component analysis (PCA), harmony integration, and dimension reduction (UMAP). Notably, the feature selection step that typically precedes PCA is skipped here, as the Xenium experiment includes only a curated selection of \(\sim300\) targets by design.
The spatialCluster() function clusters the spots, and adds the predicted cluster labels to the object. The authors recommend running with at least 10,000 iterations (nrep=1e4); we use fewer iterations in this demo for the sake of runtime. (Note that a random seed must be set (set.seed()) for the results to be reproducible.)
From here on out, both datasets could be analyzed together and/or independently, e.g., in order to identify cluster markers, annotate cell subpopulations etc.
40.7 Appendix
TipFurther reading
For further literature and resources on cross-modal analyses, we refer readers to:
Janesick, Amanda, Robert Shelansky, Andrew D. Gottscho, et al. 2023. “High Resolution Mapping of the Tumor Microenvironment Using Integrated Single-Cell, Spatial and in Situ Analysis.”Nature Communications 14 (8353). https://doi.org/10.1038/s41467-023-43458-x.