30 Differential spatial patterns
30.1 Preamble
30.1.1 Introduction
Recent advances in spatial transcriptomics (ST) techniques have facilitated the generation of multi-sample datasets across various experimental conditions (e.g. healthy vs. diseased, or multiple treatments).
When multi-sample, multi-condition ST data are available, differential analysis can be performed to identify genes with distinct spatial expression patterns that change between conditions either across domains or within any domain; we call these differential spatial pattern (DSP) genes. A DSP gene may be spatially variable in one condition and display spatially uniform expression in another one, or it could be spatially variable in all conditions, but with distinct spatial expression patterns. Conversely, genes that show spatially uniform abundance, or that are spatially variable with the same spatial structure in all conditions, are not considered DSP, because their spatial distribution is unchanged. Another way to think about DSP is that it is the spatial analog of ‘differential state analysis’ for single cell analyses. That is, here we focus on differences in expression within any domain (or different across domains) between conditions.
Identifying DSP genes can provide valuable insights into how gene expression spatial structures change across experimental conditions, helping to understand the underlying biological processes.
30.1.2 Dependencies
30.1.3 Load data
In this demo, we will analyze a Stereo-seq dataset from axolotl brain tissues collected at various stages of regeneration (Wei et al. 2022), which includes multiple samples (i.e. serial sections) measured under multiple experimental conditions (regeneration stages).
In the original dataset, which is publicly available through the Spatial Transcript Omics DataBase, there are 5 stages with multiple sections, totaling 16 samples (3 to 4 sections per stage).
In this chapter, for computational reasons, we use 3 stages – 2, 10, and 20 days post injury (DPI) – and only two sections per stage. The analysis framework uses pre-computed spatial domains generated with Banksy (Singhal et al. 2024). For performing clustering with Banksy, refer to Chapter 23. For preprocessing details of this dataset, see the muSpaData Bioconductor package.
Code
spe <- Wei22_example()Code
colnames(spatialCoords(spe)) <- c("x", "y")
plotCoords(spe,
annotate = "Banksy_smooth",
in_tissue = NULL,
x_coord = "sdimx",
y_coord = "sdimy",
y_reverse = FALSE,
sample_id = "sample_id") +
theme(legend.key.size = unit(0, "lines")) +
scale_color_manual(values=unname(pals::trubetskoy()))
30.2 DESpace
Here, we briefly demonstrate how to identify DSP genes using the DESpace (Cai, Robinson, and Tiberi 2024) Bioconductor package. For more details, see the package vignette. In brief, spot- or cell-level counts are aggregated into pseudobulk counts for each sample and domain, and these are compared across domains and conditions. DSP focuses on testing whether (condition x domain) interaction terms in the model are different from zero. In other words, does the expression of a gene change between conditions, but change differently for some domains compared to others?
Code
# run DESpace
dsp <- dsp_test(spe = spe, cluster_col = "Banksy_smooth",
sample_col = "sample_id", condition_col = "condition",
verbose = TRUE)Code
head(dsp_global <- dsp$gene_results, n = 3)## gene_id logFC.condition20DPI.cluster_id1
## AMEX60DD014721 AMEX60DD014721 -0.07170723
## AMEX60DD045083 AMEX60DD045083 0.08759059
## AMEX60DD011151 AMEX60DD011151 -0.97377297
## logFC.condition2DPI.cluster_id1
## AMEX60DD014721 -0.3898784
## AMEX60DD045083 0.5418637
## AMEX60DD011151 -2.1019048
## logFC.condition20DPI.cluster_id2
## AMEX60DD014721 -0.8510223
## AMEX60DD045083 -1.3377605
## AMEX60DD011151 0.1861520
## logFC.condition2DPI.cluster_id2
## AMEX60DD014721 0.8526301
## AMEX60DD045083 1.1226997
## AMEX60DD011151 0.4671347
## logFC.condition20DPI.cluster_id3
## AMEX60DD014721 -0.8328237
## AMEX60DD045083 0.2478193
## AMEX60DD011151 -0.4011555
## logFC.condition2DPI.cluster_id3
## AMEX60DD014721 -0.9450830
## AMEX60DD045083 1.2666344
## AMEX60DD011151 0.4086612
## logFC.condition20DPI.cluster_id4
## AMEX60DD014721 0.22287797
## AMEX60DD045083 -0.99648014
## AMEX60DD011151 -0.01634095
## logFC.condition2DPI.cluster_id4 logCPM LR
## AMEX60DD014721 0.2085777 9.344907 100.69288
## AMEX60DD045083 -0.7969334 7.505402 97.72697
## AMEX60DD011151 0.1810959 7.959492 85.69149
## PValue FDR
## AMEX60DD014721 3.081052e-18 1.540526e-14
## AMEX60DD045083 1.243325e-17 3.108313e-14
## AMEX60DD011151 3.472538e-15 5.787563e-12Code
# count significant DSP genes (at 5% FDR significance level)
table(dsp_global$FDR <= 0.05)##
## FALSE TRUE
## 4809 191In order to identify the key spatial cluster (domain) where expression changes across conditions, we use the individual_dsp() function, which focuses on domain-specific DSP.
Code
dsp_clu <- individual_dsp(spe, cluster_col = "Banksy_smooth",
sample_col = "sample_id", condition_col = "condition")Code
# results for cluster 2
dsp_clu2 <- dsp_clu$`2`
head(dsp_clu2, n = 3)## gene_id logFC.condition20DPI.cluster_id2
## AMEX60DD014721 AMEX60DD014721 -0.5801351
## AMEX60DD014991 AMEX60DD014991 2.6944195
## AMEX60DD055246 AMEX60DD055246 -0.2660938
## logFC.condition2DPI.cluster_id2 logCPM LR
## AMEX60DD014721 1.270051 9.582333 74.62550
## AMEX60DD014991 3.015258 7.330327 73.65886
## AMEX60DD055246 -2.298605 5.661946 61.49402
## PValue FDR
## AMEX60DD014721 6.241352e-17 2.530002e-13
## AMEX60DD014991 1.012001e-16 2.530002e-13
## AMEX60DD055246 4.433456e-14 7.389093e-11Code
# top DSP gene for cluster 2
top_dsp <- dsp_clu2$gene_id[1]
# get gene symbols from Ensembl identifiers
idx <- match(top_dsp, rowData(spe)$gene_name)
(.gs <- rowData(spe)$gene_id[idx])## [1] "ECM1"30.3 Visualization
DSP genes can be further investigated, for example, by plotting the spatial expression of genes across conditions, or the log2-transformed counts per million of genes across different clusters and conditions.
Code
.ids <- levels(spe$sample_id)
ps <- lapply(seq_along(.ids), \(.) {
spe_j <- spe[, spe$sample_id == .ids[.]]
p <- FeaturePlot(spe_j,
feature = top_dsp,
platform = "Stereo-seq",
coordinates = c("sdimx", "sdimy"),
cluster_col = "Banksy_smooth",
cluster = '2',
diverging = TRUE,
low = "gray95",
high = "blue") +
ggtitle(.ids[.])
# ignore alignment
free(p[[1]])
})
wrap_plots(ps, ncol=3) & theme(
plot.title = element_text(margin = margin(b = 100), hjust = 0.5))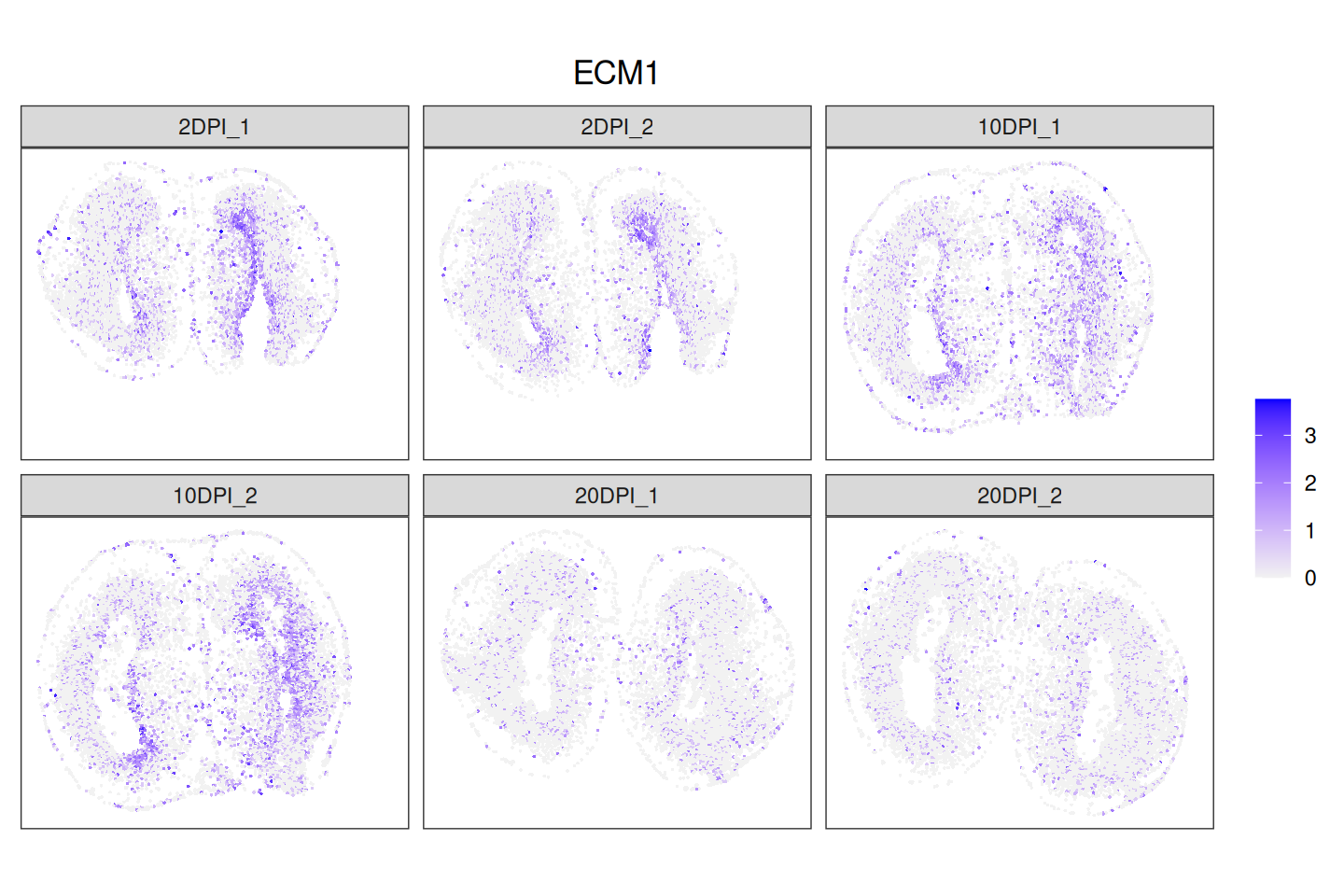
Code
# calculate log CPM
cps <- edgeR::cpm(dsp$estimated_y, log = TRUE)
cps_name <- colnames(cps)
mdata <- data.frame(
log_cpm = cps[top_dsp, ],
Banksy_smooth = factor(sub(".*_", "", cps_name)),
day = as.numeric(sub("([0-9]+)DPI.*", "\\1", cps_name)),
sample_id = sub("(_[0-9]+)$", "", cps_name)
)
ggplot(mdata, aes(x = factor(day), y = log_cpm)) +
geom_jitter(aes(color = Banksy_smooth), size = 2, width = 0.1) +
geom_boxplot(aes(fill = ifelse(Banksy_smooth == "2",
"cluster 2", "non-cluster 2"))) +
scale_x_discrete(breaks = c(2, 10, 20)) +
scale_fill_manual(values = c("#4DAF4A", "gray")) +
labs(title = .gs, x = "Days post injury",
y = "log2 counts per million (logCPM)", fill = "",
color = "Banksy cluster") +
theme(legend.position = "right")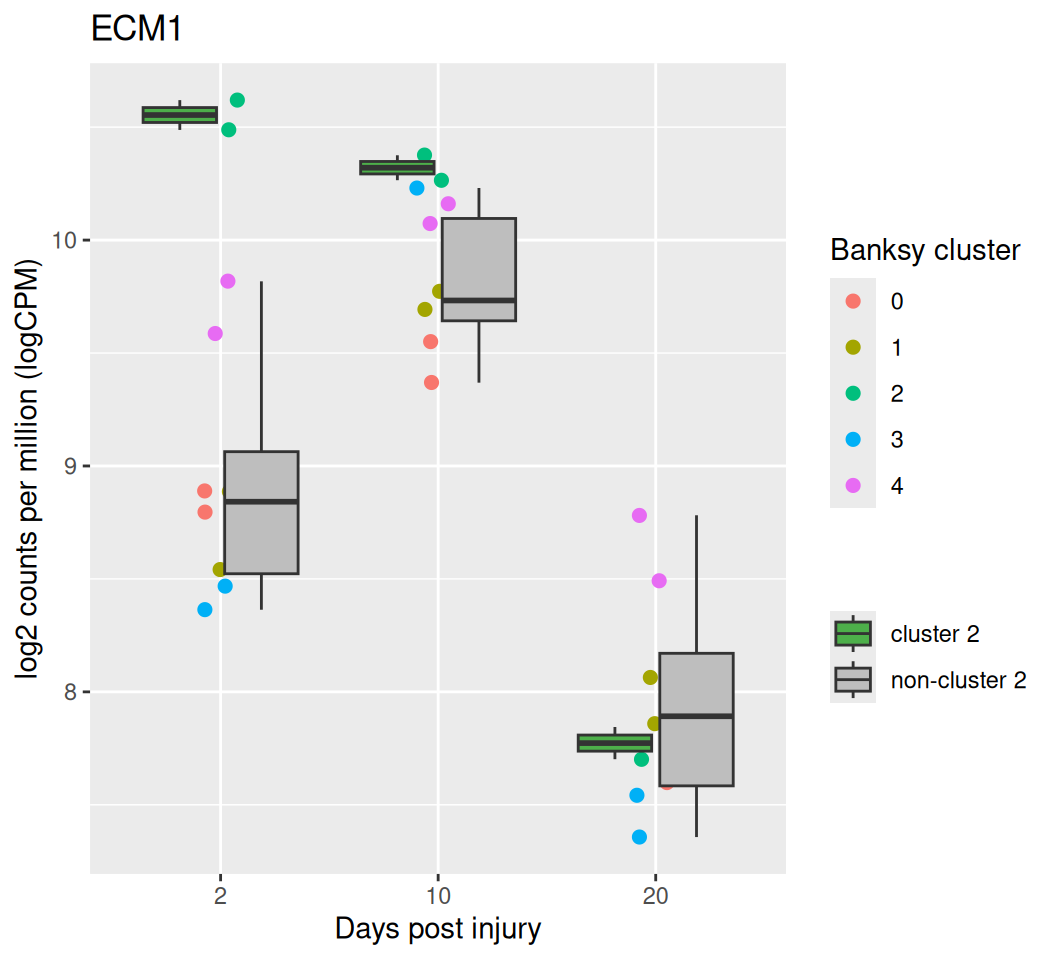
The boxplots above show the average log-CPM for cluster 2 and for all other clusters (excluding cluster 2) across different time points. The largest difference between cluster 2 and the remaining clusters is observed at 2 DPI, with the difference decreasing over time. At 20 DPI, no significant log-CPM differences are observed between the clusters. This suggests that spatial patterns change across conditions (i.e. time stages).