12 Workflow: Visium DLPFC
12.1 Preamble
12.1.1 Introduction
This workflow analyzes a 10x Genomics Visium dataset consisting of one sample (Visium capture area) of postmortem human brain tissue from the dorsolateral prefrontal cortex (DLPFC) region, originally described by Maynard et al. (2021).
The original full dataset contains 12 samples in total, from 3 donors, with 2 pairs of spatially adjacent replicates (serial sections) per donor (4 samples per donor). Each sample spans several cortical layers plus white matter in a tissue section. The examples in this workflow use a single representative sample, labeled 151673, which is often illustrated in various method papers in spatial omics data analysis.
For more details on the dataset, see Maynard et al. (2021). The full dataset is publicly available through the spatialLIBD Bioconductor package (Pardo et al. 2022). The dataset can also be explored interactively through the spatialLIBD Shiny web app.
12.1.2 Dependencies
12.2 Workflow
12.2.1 Load data
Load sample 151673 from the DLPFC dataset. This sample is available as a SpatialExperiment object from the STexampleData package.
Code
# load data
spe <- Visium_humanDLPFC()
class(spe)## [1] "SpatialExperiment"
## attr(,"package")
## [1] "SpatialExperiment"Code
dim(spe)## [1] 33538 499212.2.2 Plot data
As an initial check, plot the spatial coordinates (spots) in x-y dimensions, to check that the object has loaded correctly. We use plotting functions from the ggspavis package.
Code
# plot spatial coordinates (spots)
plotCoords(spe)
12.2.3 Quality control (QC)
We calculate quality control (QC) metrics using the scater package (McCarthy et al. 2017), and apply simple global thresholding-based QC methods to identify any low-quality spots, as described in Section 9.3. More details, including more advanced QC approaches, are described in Chapter 9.
Code
# subset to keep only spots over tissue
spe <- spe[, spe$in_tissue == 1]
dim(spe)## [1] 33538 3639Code
## is_mito
## FALSE TRUE
## 33525 13Code
rowData(spe)$gene_name[is_mito]## [1] "MT-ND1" "MT-ND2" "MT-CO1" "MT-CO2" "MT-ATP8" "MT-ATP6" "MT-CO3"
## [8] "MT-ND3" "MT-ND4L" "MT-ND4" "MT-ND5" "MT-ND6" "MT-CYB"Calculate QC metrics using scater (McCarthy et al. 2017).
Code
# calculate per-spot QC metrics and store in colData
spe <- addPerCellQC(spe, subsets = list(mito = is_mito))
head(colData(spe), 3)## DataFrame with 3 rows and 14 columns
## barcode_id sample_id in_tissue array_row
## <character> <character> <integer> <integer>
## AAACAAGTATCTCCCA-1 AAACAAGTATCTCCCA-1 sample_151673 1 50
## AAACAATCTACTAGCA-1 AAACAATCTACTAGCA-1 sample_151673 1 3
## AAACACCAATAACTGC-1 AAACACCAATAACTGC-1 sample_151673 1 59
## array_col ground_truth reference cell_count sum
## <integer> <character> <character> <integer> <numeric>
## AAACAAGTATCTCCCA-1 102 Layer3 Layer3 6 8458
## AAACAATCTACTAGCA-1 43 Layer1 Layer1 16 1667
## AAACACCAATAACTGC-1 19 WM WM 5 3769
## detected subsets_mito_sum subsets_mito_detected
## <integer> <numeric> <integer>
## AAACAAGTATCTCCCA-1 3586 1407 13
## AAACAATCTACTAGCA-1 1150 204 11
## AAACACCAATAACTGC-1 1960 430 13
## subsets_mito_percent total
## <numeric> <numeric>
## AAACAAGTATCTCCCA-1 16.6351 8458
## AAACAATCTACTAGCA-1 12.2376 1667
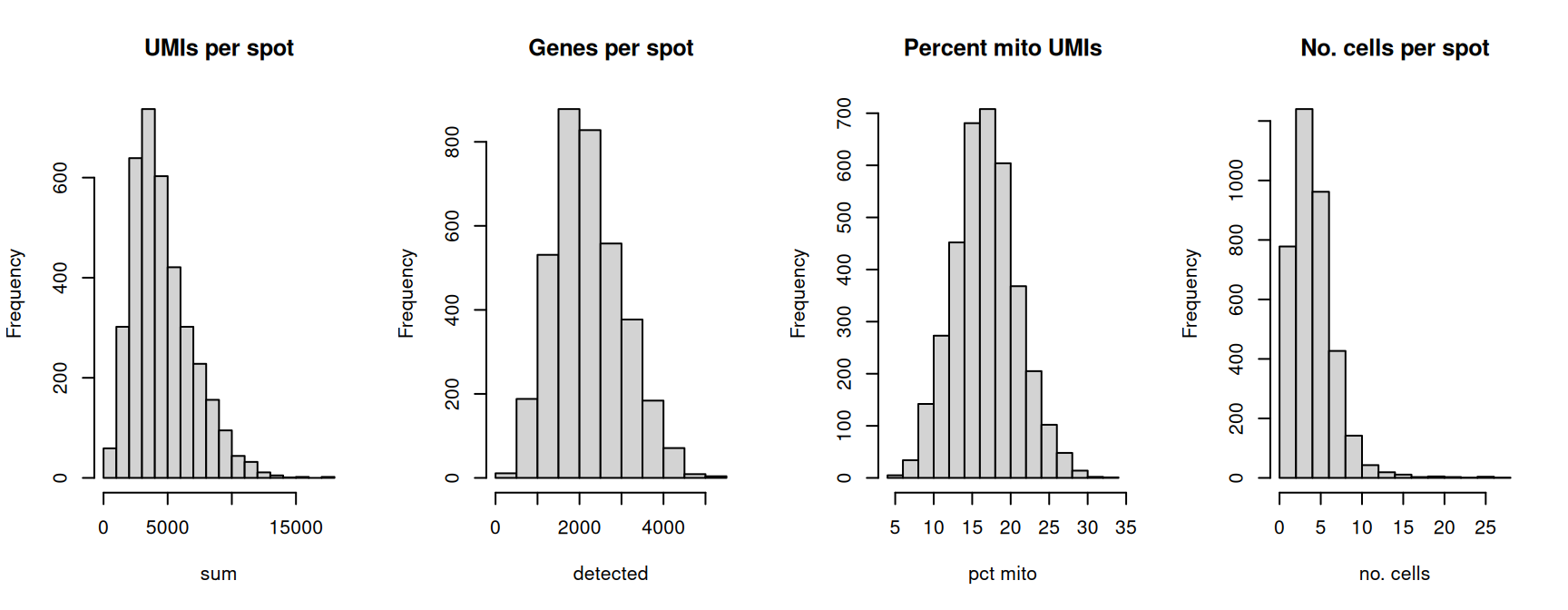
## AAACACCAATAACTGC-1 11.4089 3769Select global filtering thresholds for the QC metrics by examining distributions using histograms.
Code
Code
# select global QC thresholds
spe$qc_lib_size <- spe$sum < 600
spe$qc_detected <- spe$detected < 400
spe$qc_mito <- spe$subsets_mito_percent > 28
table(spe$qc_lib_size)##
## FALSE TRUE
## 3631 8Code
table(spe$qc_detected)##
## FALSE TRUE
## 3632 7Code
table(spe$qc_mito)##
## FALSE TRUE
## 3622 17Plot the spatial distributions of the potentially identified low-quality spots, to ensure that they are not concentrated within biologically meaningful regions (which could suggest that the selected thresholds were too stringent).
Code
# plot spatial distributions of discarded spots
p1 <- plotObsQC(spe,
plot_type = "spot",
annotate = "qc_lib_size") +
ggtitle("Library size (< threshold)")
p2 <- plotObsQC(spe,
plot_type = "spot",
annotate = "qc_detected") +
ggtitle("Detected genes (< threshold)")
p3 <- plotObsQC(spe,
plot_type = "spot",
annotate = "qc_mito") +
ggtitle("Mito proportion (> threshold)")
# arrange plots using patchwork package
p1 | p2 | p3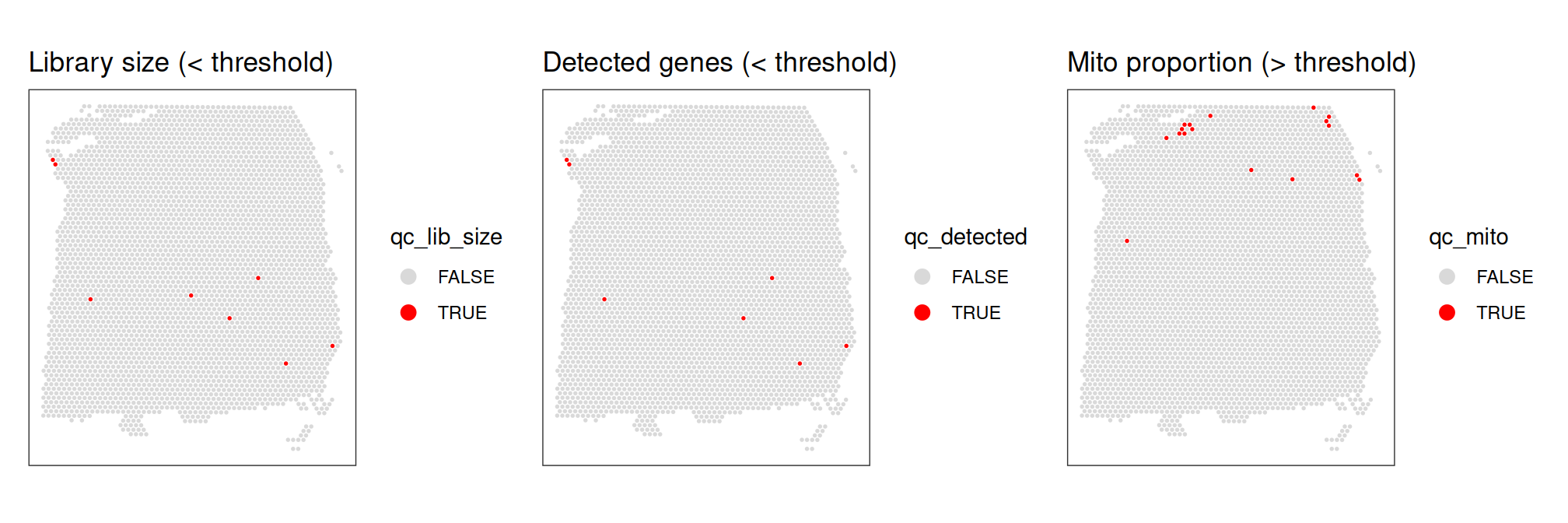
Select spots to discard by combining the sets of identified low-quality spots according to each metric.
Code
## [1] 8 7 17Code
# combined set of identified spots
spe$discard <- spe$qc_lib_size | spe$qc_detected | spe$qc_mito
table(spe$discard)##
## FALSE TRUE
## 3614 25Plot the spatial distribution of the combined set of identified low-quality spots to discard, to again confirm that they do not correspond to any clearly biologically meaningful regions, which could indicate that we are removing biologically informative spots. Specifically, in this dataset, we want to ensure that the discarded spots do not correspond to a single cortical layer.
Code
# check spatial pattern of discarded spots
plotObsQC(spe, plot_type = "spot", annotate = "discard")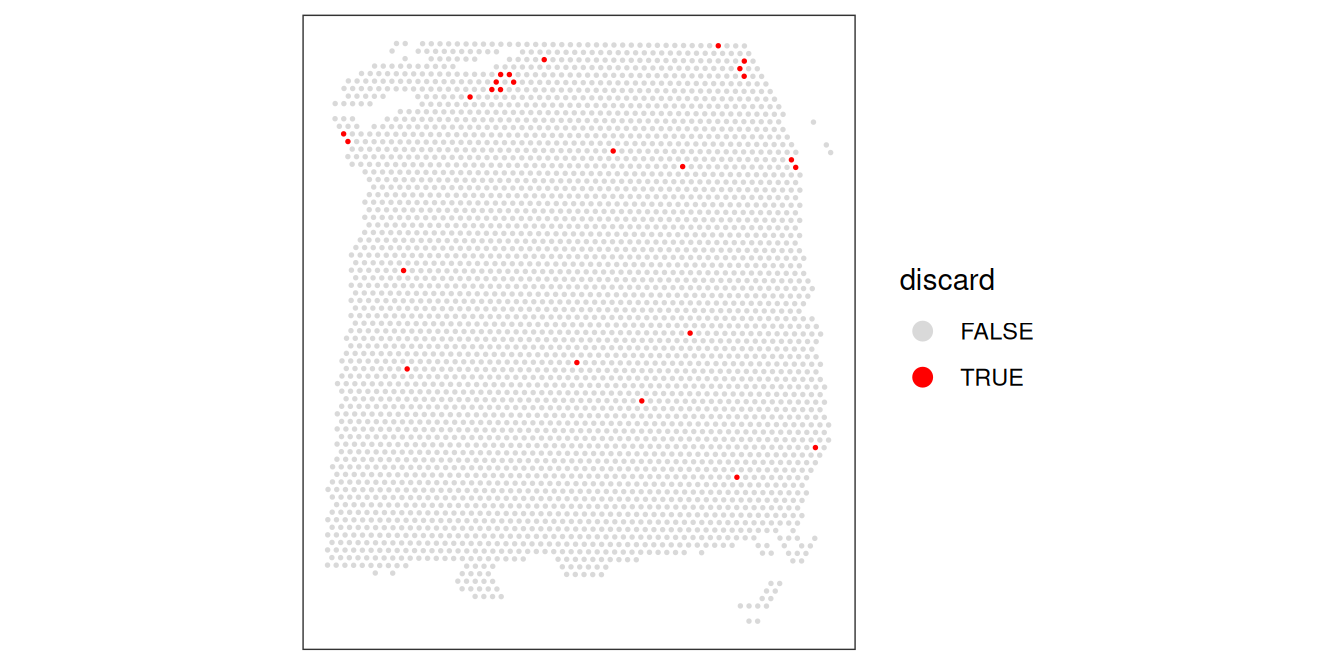
Filter out the low-quality spots.
Code
# filter out low-quality spots
spe <- spe[, !spe$discard]
dim(spe)## [1] 33538 361412.2.4 Normalization
Calculate log-transformed normalized counts (logcounts) using library size normalization, as described in Section 10.2. We use methods from the scater (McCarthy et al. 2017) and scran (Lun, McCarthy, and Marioni 2016) packages, making the simplified assumption that spots can be treated as equivalent to single cells. For more details and other options, see Chapter 10.
Code
# calculate library size factors
spe <- computeLibraryFactors(spe)
summary(sizeFactors(spe))## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.1330 0.6329 0.8978 1.0000 1.2872 3.7820Code
hist(sizeFactors(spe), breaks = 20, main = "Histogram of size factors")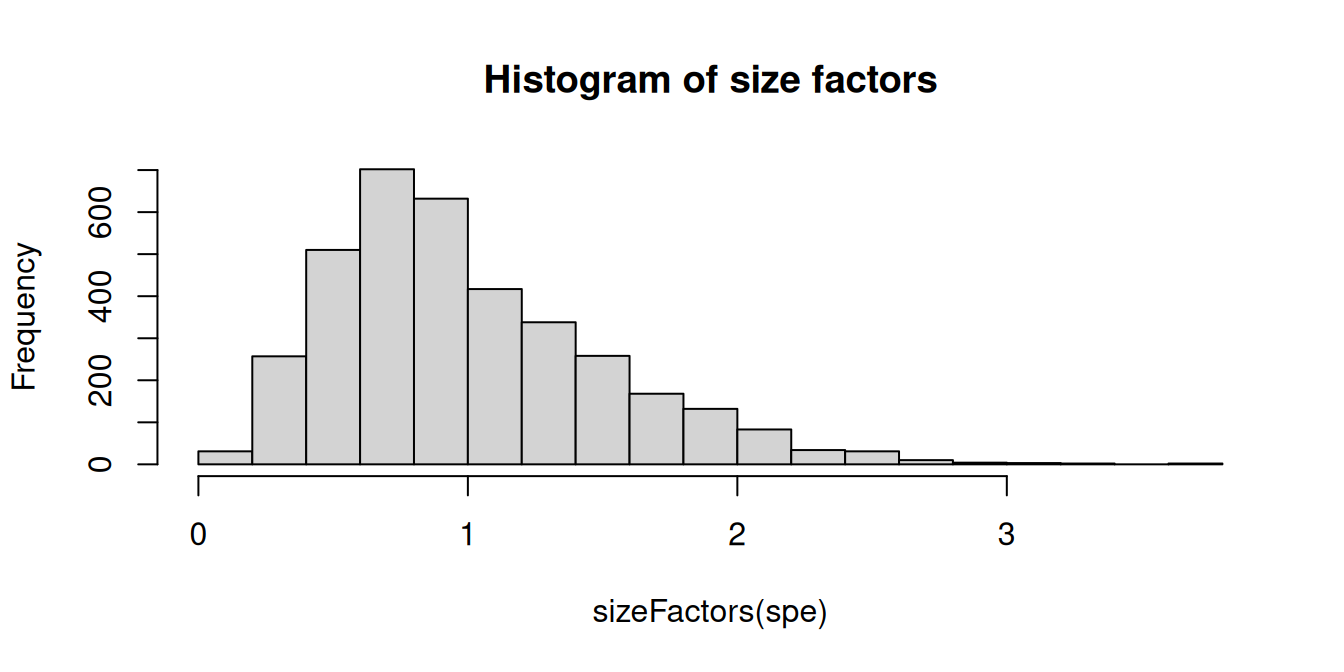
Code
# calculate logcounts
spe <- logNormCounts(spe)
assayNames(spe)## [1] "counts" "logcounts"12.2.5 Feature selection (HVGs)
Apply feature selection methods to identify a set of top highly variable genes (HVGs). We use methods from the scran (Lun, McCarthy, and Marioni 2016) package, again making the simplified assumption that spots can be treated as equivalent to single cells. We also first remove mitochondrial genes, since these tend to be very highly expressed and are not of main biological interest. For more details, see Chapter 10.
For details on alternative feature selection methods to identify spatially variable genes (SVGs) instead of HVGs, for example using the nnSVG (Weber et al. 2023) or other packages, see Section 24.3.1.
Code
# remove mitochondrial genes
spe <- spe[!is_mito, ]
dim(spe)## [1] 33525 3614Code
# fit mean-variance relationship, decomposing variance into
# technical and biological components
dec <- modelGeneVar(spe)
# select top HVGs
top_hvgs <- getTopHVGs(dec, prop = 0.1)
# number of HVGs selected
length(top_hvgs)## [1] 142412.2.6 Dimensionality reduction
Next, we perform dimensionality reduction using principal component analysis (PCA), applied to the set of top HVGs. We retain the top 50 principal components (PCs) for further downstream analyses. This is done both to reduce noise and to improve computational efficiency. We also run UMAP on the set of top 50 PCs and retain the top 2 UMAP components for visualization purposes.
We use the computationally efficient implementation of PCA from the scater package (McCarthy et al. 2017), which uses randomization and therefore requires setting a random seed for reproducibility.
See Chapter 10 and Chapter 22 for more details.
Code
# using scater package
set.seed(123)
spe <- runPCA(spe, subset_row = top_hvgs)
spe <- runUMAP(spe, dimred = "PCA")
reducedDimNames(spe)## [1] "PCA" "UMAP"Code
dim(reducedDim(spe, "PCA"))## [1] 3614 50Code
dim(reducedDim(spe, "UMAP"))## [1] 3614 2Code
# update column names for plotting
colnames(reducedDim(spe, "UMAP")) <- paste0("UMAP", 1:2)12.2.7 Clustering
Next, we apply a clustering algorithm to identify cell types or spatial domains. Note that we are using only molecular features (gene expression) as the input for clustering in this example. Alternatively, we could use a spatially-aware clustering algorithm, as demonstrated in the example in Section 10.5.
Here, we use graph-based clustering using the Walktrap method implemented in scran, applied to the top 50 PCs calculated on the set of top HVGs from above.
For more details on clustering, see Chapter 23.
Code
# graph-based clustering
set.seed(123)
k <- 10
g <- buildSNNGraph(spe, k = k, use.dimred = "PCA")
g_walk <- igraph::cluster_walktrap(g)
clus <- g_walk$membership
table(clus)## clus
## 1 2 3 4 5 6 7
## 365 765 875 803 198 370 238Visualize the cluster labels by plotting in x-y space, alongside the manually annotated reference labels (ground_truth) available for this dataset.
Code
# plot cluster labels in x-y space
plotCoords(spe, annotate = "label", pal = "libd_layer_colors")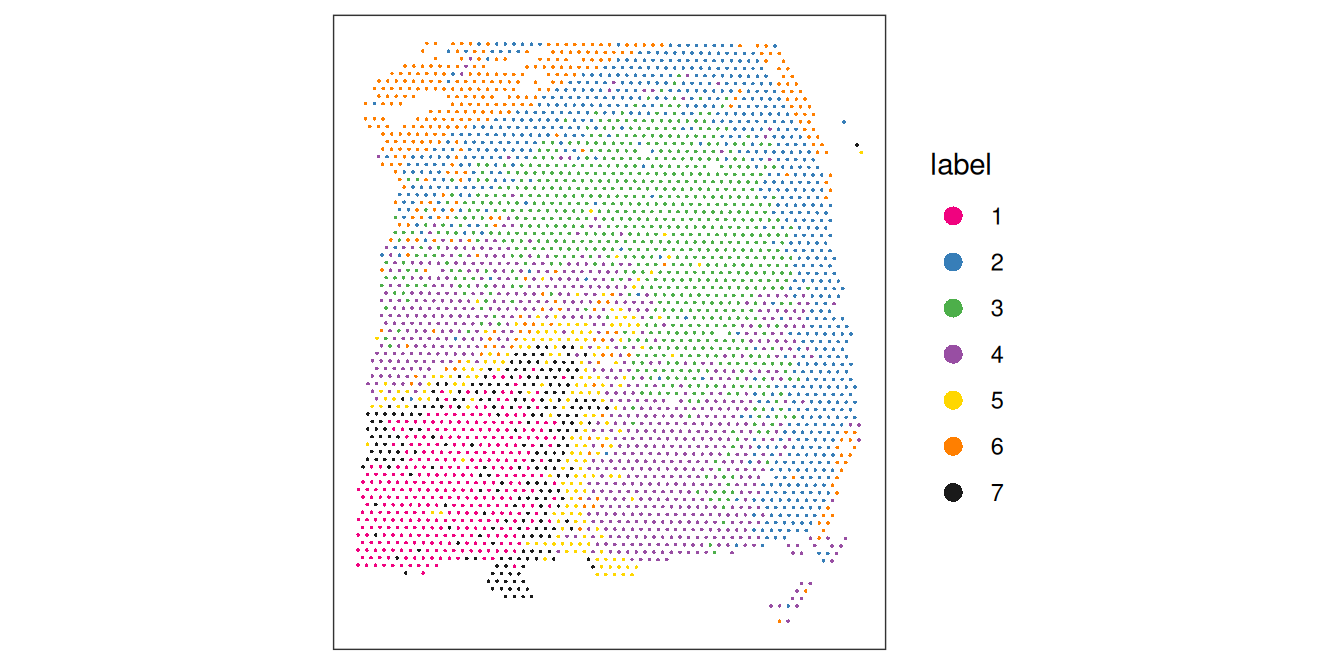
Code
# plot manually annotated reference labels
plotCoords(spe, annotate = "ground_truth", pal = "libd_layer_colors")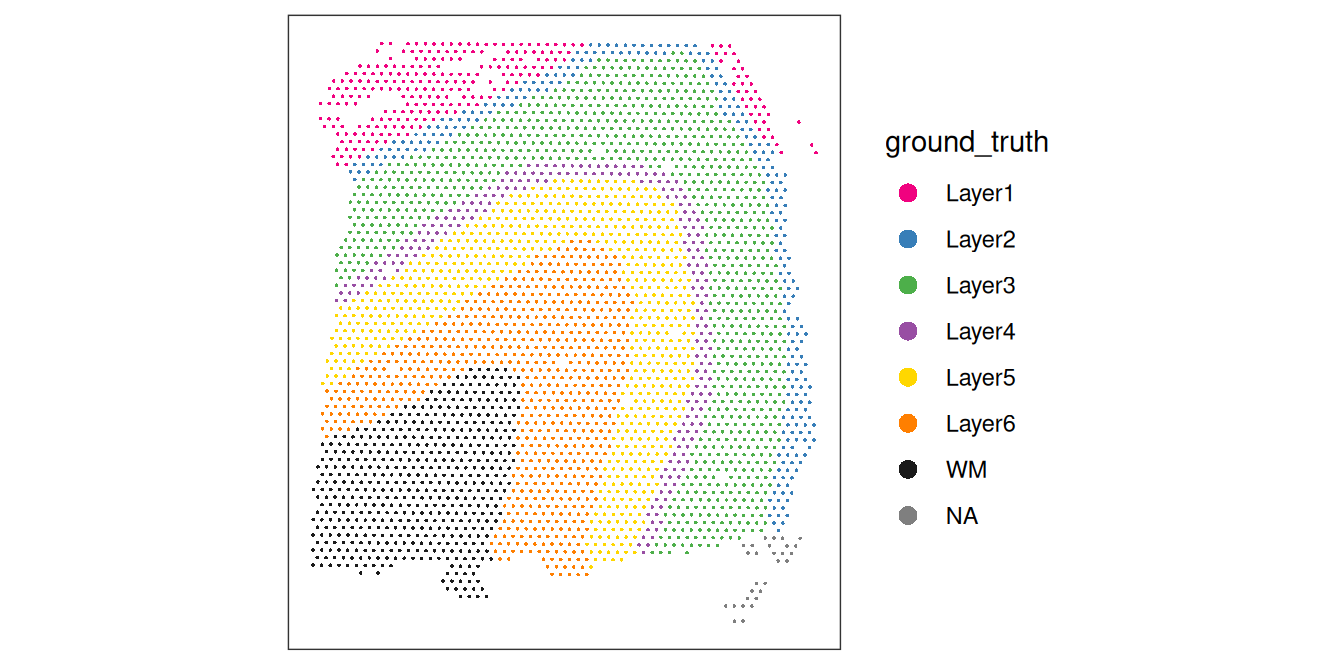
We can also plot the cluster labels in the top 2 UMAP dimensions.
Code
# plot clusters labels in UMAP dimensions
plotDimRed(spe, plot_type = "UMAP", annotate = "label",
pal = "libd_layer_colors")
12.2.8 Differential expression
Identify marker genes for each cluster or spatial domain by testing for differentially expressed genes using pairwise t-tests, specifically testing for upregulation for each cluster or spatial domain.
We use the scran package (Lun, McCarthy, and Marioni 2016) to calculate the differential tests. We use a binomial test, which is a more stringent test than the default pairwise t-tests, and tends to select genes that are easier to interpret and validate experimentally.
See Chapter 10 or Chapter 23 for more details.
Code
## [1] 49Visualize the marker genes using a heatmap.
Code
pbs <- aggregateAcrossCells(spe,
ids = spe$label, subset.row = top,
use.assay.type = "logcounts", statistics = "mean")
# use gene symbols as feature names
mtx <- t(assay(pbs))
colnames(mtx) <- rowData(pbs)$gene_name
# plot using pheatmap package
pheatmap(mat = mtx, scale = "column")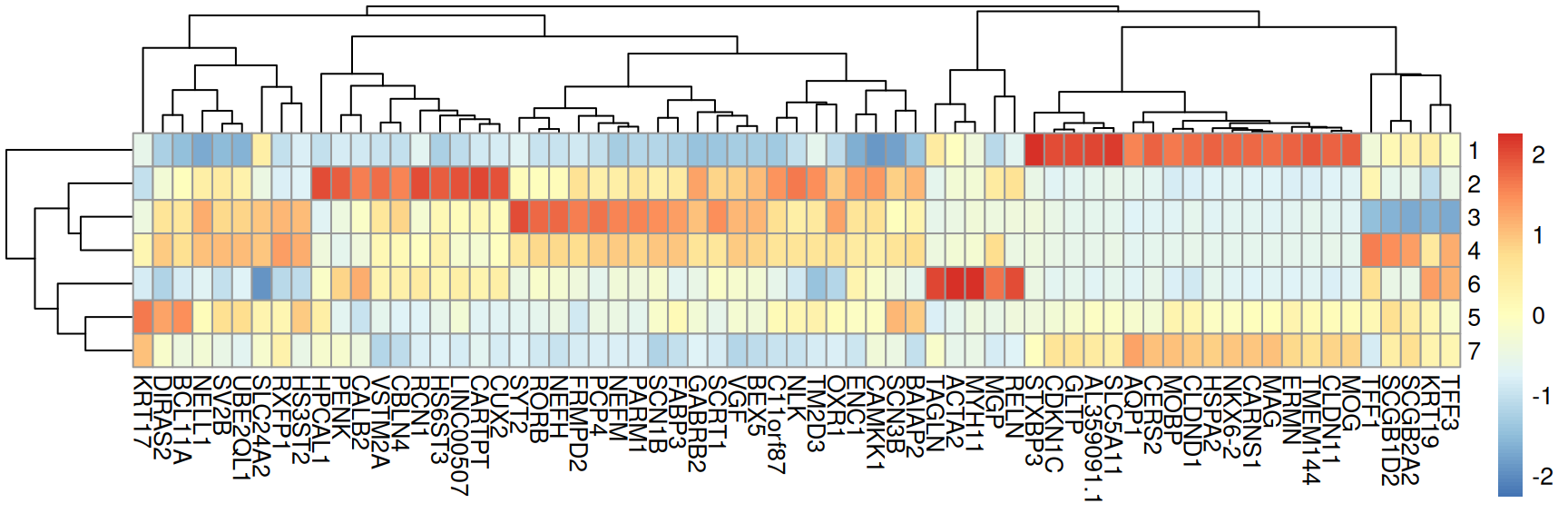
12.3 spatialLIBD
The examples above demonstrated a streamlined analysis workflow for the Visium DLPFC dataset (Maynard et al. 2021). In this section, we will use the spatialLIBD package (Pardo et al. 2022) to continue analyzing this dataset by creating an interactive Shiny website to visualize the data.
12.3.1 Why use spatialLIBD?
The spatialLIBD package has a function, spatialLIBD::run_app(spe), which will create an interactive website using a SpatialExperiment object (spe). The interactive website it creates has several features that were initially designed for the DLPFC dataset (Maynard et al. 2021) and later made flexible for any dataset (Pardo et al. 2022). These features include panels to visualize Visium spots:
- for one tissue section at a time, either with interactive or static versions
- for multiple tissue sections at a time, either interactively or statically
Both options work with continuous and discrete variables such as the gene expression and clusters, respectively. The interactive version for discrete variables such as clusters is useful if you want to manually annotate Visium spots, as in Maynard et al. (2021). spatialLIBD allows users to download the annotated spots and resume your spot annotation work later.

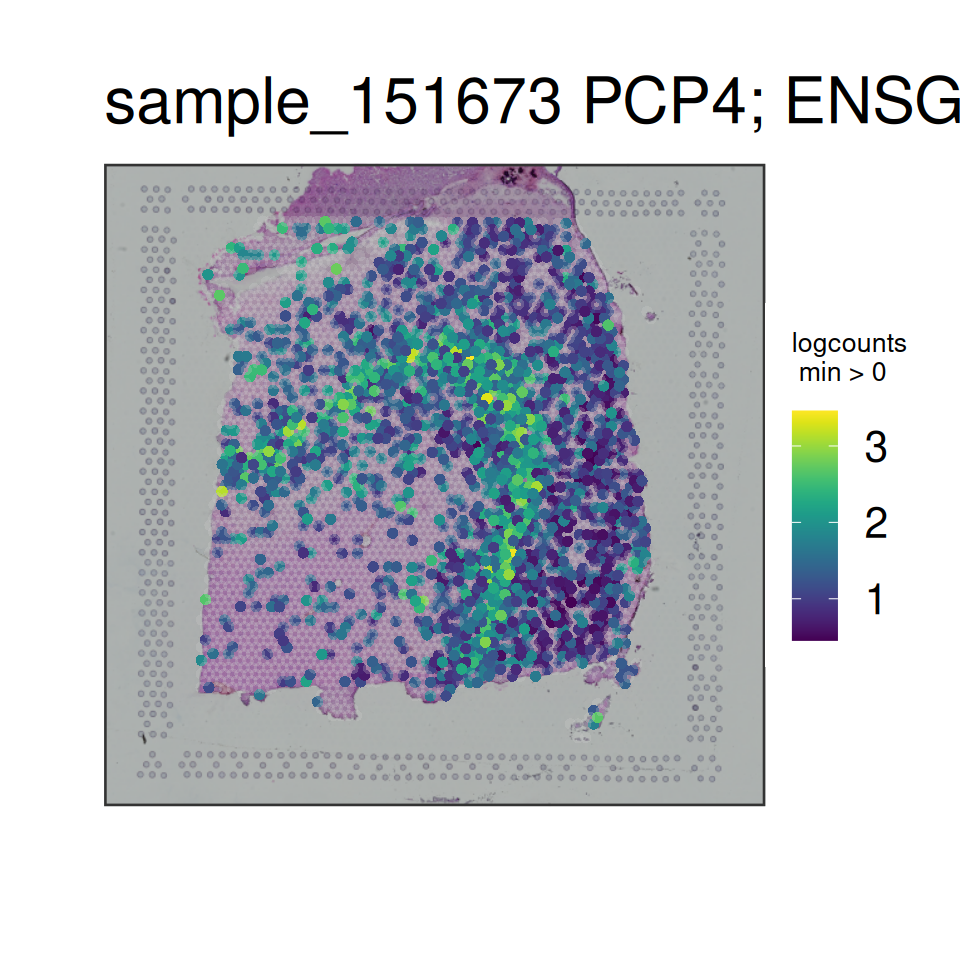
spatialLIBD::run_app(spatialLIBD::fetch_data('spe')) version 1.4.0 and then using the lasso selection, we selected a set of spots in the UMAP interactive plot colored by the estimated number of cells per spot (cell_count) on the bottom left, which automatically updated the other three plots.Visualizing genes or clusters across multiple tissue sections can be useful. For example, here we show the expression levels of PCP4 across two sets of spatially adjacent replicates. PCP4 is a marker gene for layer 5 in the gray matter of the DLPFC in the human brain. Spatially adjacent replicates are about 10 μm apart from each other and visualizations like the one below help assess the technical variability in the Visium technology.

spatialLIBD::run_app(spatialLIBD::fetch_data('spe')) version 1.4.0, selecting the PCP4 gene, selecting the paper gene color scale, changing the number of rows and columns in the grid 2, selecting two pairs of spatially adjacent replicate samples (151507, 151508, 151673, and 151674), and clicking on the upgrade grid plot button. Note that the default viridis gene color scale is color-blind friendly.You can try out a spatialLIBD-powered website yourself by opening it on your browser.
shiny-powered websites work best on browsers such as Google Chrome and Mozilla Firefox, among others.12.3.2 Want to learn more about spatialLIBD?
For more details about spatialLIBD, please check the spatialLIBD Bioconductor landing page or the pkgdown documentation website. In particular, we have two vignettes:
You can also read more about spatialLIBD in the associated publication Pardo et al. (2022).
If you prefer to watch videos, recorded presentations related to the dataset (Maynard et al. 2021) and spatialLIBD (Pardo et al. 2022) are also available here.
12.3.3 Code prerequisites
For this demo, we will re-use the spe object (in SpatialExperiment format) created in the example DLPFC workflow above. If you are starting from here, you can re-build the object by running the code in section Section 12.2 above.
We also load some additional dependency packages, in addition to the dependencies already loaded in Section 12.2 above.
In addition, we will modify the final step of the workflow above, where we identified marker genes per cluster by differential expression testing. We will modify this step to summarize the results of the differential expression testing in a different way, and store this information in the spe object.
12.3.4 Prepare for spatialLIBD
We also need to modify the spe object, similar to steps we need to carry out when using spatialLIBD with 10x Genomics public datasets.
12.3.4.1 Basic information
12.3.4.2 Gene annotation
Since the gene information is missing, we will add gene annotation data from Gencode. Alternatively, ideally you would add this information from the same gene annotation used for originally running Space Ranger.
Code
# download Gencode v32 GTF file and cache it
bfc <- BiocFileCache::BiocFileCache()
gtf_cache <- BiocFileCache::bfcrpath(
bfc,
paste0(
"ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/",
"release_32/gencode.v32.annotation.gtf.gz"
)
)## adding rname 'ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gtf.gz'Code
# show GTF cache location
gtf_cache## BFC8
## "/root/.cache/R/BiocFileCache/46a3252e0146_gencode.v32.annotation.gtf.gz"Code
# import into R (takes ~1 min)
gtf <- rtracklayer::import(gtf_cache)
# subset to genes only
gtf <- gtf[gtf$type == "gene"]
# remove the .x part of the gene IDs
gtf$gene_id <- gsub("\\..*", "", gtf$gene_id)
# set the names to be the gene IDs
names(gtf) <- gtf$gene_id
# match the genes
match_genes <- match(rowData(spe)$gene_id, gtf$gene_id)
table(is.na(match_genes))##
## FALSE TRUE
## 33267 258Code
# drop the few genes for which we don't have information
spe <- spe[!is.na(match_genes), ]
match_genes <- match_genes[!is.na(match_genes)]
# keep only some columns from the gtf
mcols(gtf) <- mcols(gtf)[, c("source", "type", "gene_id", "gene_name", "gene_type")]
# save the "interesting" columns from our original spe object
interesting <- rowData(spe)[, grepl("interest", colnames(rowData(spe)))]
# add gene info to spe object
rowRanges(spe) <- gtf[match_genes]
# add back the "interesting" columns
rowData(spe) <- cbind(rowData(spe), interesting)
# inspect the gene annotation data we added
rowRanges(spe)## GRanges object with 33267 ranges and 12 metadata columns:
## seqnames ranges strand | source type
## <Rle> <IRanges> <Rle> | <factor> <factor>
## ENSG00000243485 chr1 29554-31109 + | HAVANA gene
## ENSG00000237613 chr1 34554-36081 - | HAVANA gene
## ENSG00000186092 chr1 65419-71585 + | HAVANA gene
## ENSG00000238009 chr1 89295-133723 - | HAVANA gene
## ENSG00000239945 chr1 89551-91105 - | HAVANA gene
## ... ... ... ... . ... ...
## ENSG00000160298 chr21 46300181-46323875 - | HAVANA gene
## ENSG00000160299 chr21 46324141-46445769 + | HAVANA gene
## ENSG00000160305 chr21 46458891-46570015 + | HAVANA gene
## ENSG00000160307 chr21 46598604-46605208 - | HAVANA gene
## ENSG00000160310 chr21 46635595-46665124 + | HAVANA gene
## gene_id gene_name gene_type gene_interest_1
## <character> <character> <character> <logical>
## ENSG00000243485 ENSG00000243485 MIR1302-2HG lncRNA TRUE
## ENSG00000237613 ENSG00000237613 FAM138A lncRNA TRUE
## ENSG00000186092 ENSG00000186092 OR4F5 protein_coding TRUE
## ENSG00000238009 ENSG00000238009 AL627309.1 lncRNA TRUE
## ENSG00000239945 ENSG00000239945 AL627309.3 lncRNA TRUE
## ... ... ... ... ...
## ENSG00000160298 ENSG00000160298 C21orf58 protein_coding FALSE
## ENSG00000160299 ENSG00000160299 PCNT protein_coding FALSE
## ENSG00000160305 ENSG00000160305 DIP2A protein_coding FALSE
## ENSG00000160307 ENSG00000160307 S100B protein_coding FALSE
## ENSG00000160310 ENSG00000160310 PRMT2 protein_coding FALSE
## gene_interest_2 gene_interest_3 gene_interest_4
## <logical> <logical> <logical>
## ENSG00000243485 TRUE TRUE TRUE
## ENSG00000237613 TRUE TRUE TRUE
## ENSG00000186092 TRUE TRUE TRUE
## ENSG00000238009 TRUE TRUE TRUE
## ENSG00000239945 TRUE TRUE TRUE
## ... ... ... ...
## ENSG00000160298 FALSE FALSE FALSE
## ENSG00000160299 FALSE FALSE FALSE
## ENSG00000160305 FALSE FALSE FALSE
## ENSG00000160307 FALSE FALSE FALSE
## ENSG00000160310 FALSE FALSE FALSE
## gene_interest_5 gene_interest_6 gene_interest_7
## <logical> <logical> <logical>
## ENSG00000243485 TRUE TRUE TRUE
## ENSG00000237613 TRUE TRUE TRUE
## ENSG00000186092 TRUE TRUE TRUE
## ENSG00000238009 TRUE TRUE TRUE
## ENSG00000239945 TRUE TRUE TRUE
## ... ... ... ...
## ENSG00000160298 FALSE FALSE FALSE
## ENSG00000160299 FALSE FALSE FALSE
## ENSG00000160305 FALSE FALSE FALSE
## ENSG00000160307 FALSE FALSE FALSE
## ENSG00000160310 FALSE FALSE FALSE
## -------
## seqinfo: 25 sequences from an unspecified genome; no seqlengthsNow that we have the gene annotation information, we can use it to add a few more pieces to our spe object that spatialLIBD will use. For example, we want to enable users to search genes by either their gene symbol or their Ensembl ID. We would also like to visualize the amount and percent of the mitochondrial gene expression.
Code
# add information used by spatialLIBD
rowData(spe)$gene_search <- paste0(
rowData(spe)$gene_name, "; ", rowData(spe)$gene_id
)
# compute chrM expression and chrM expression ratio
is_mito <- which(seqnames(spe) == "chrM")
spe$expr_chrM <- colSums(counts(spe)[is_mito, , drop = FALSE])
spe$expr_chrM_ratio <- spe$expr_chrM / spe$sum_umi12.3.4.3 Extra information and filtering
Now that we have the full gene annotation information we need, we can proceed to add some last touches as well as filter the object to reduce the memory required for visualizing the data.
Code
## [1] 11547## [1] 34.71007Code
spe <- spe[-no_expr, , drop = FALSE]
# remove spots without counts
summary(spe$sum_umi)## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 537 2400 3442 3849 4921 15862Code
# if we had spots with no counts, we would remove them
if (any(spe$sum_umi == 0)) {
spots_no_counts <- which(spe$sum_umi == 0)
# number of spots with no counts
print(length(spots_no_counts))
# percent of spots with no counts
print(length(spots_no_counts) / ncol(spe) * 100)
spe <- spe[, -spots_no_counts, drop = FALSE]
}We should now be ready to proceed to making our interactive website. To confirm, we can use the spatialLIBD::check_spe() to verify that everything is set up correctly. If not, this function will tell us what we missed.
Code
# run check_spe() function
spatialLIBD::check_spe(spe)## class: SpatialExperiment
## dim: 21720 3614
## metadata(0):
## assays(2): counts logcounts
## rownames(21720): ENSG00000243485 ENSG00000238009 ... ENSG00000160307
## ENSG00000160310
## rowData names(13): source type ... gene_interest_7 gene_search
## colnames(3614): AAACAAGTATCTCCCA-1 AAACAATCTACTAGCA-1 ...
## TTGTTTGTATTACACG-1 TTGTTTGTGTAAATTC-1
## colData names(26): barcode_id sample_id ... expr_chrM_ratio
## ManualAnnotation
## reducedDimNames(2): PCA UMAP
## mainExpName: NULL
## altExpNames(0):
## spatialCoords names(2) : pxl_col_in_fullres pxl_row_in_fullres
## imgData names(4): sample_id image_id data scaleFactor12.3.5 Explore the data
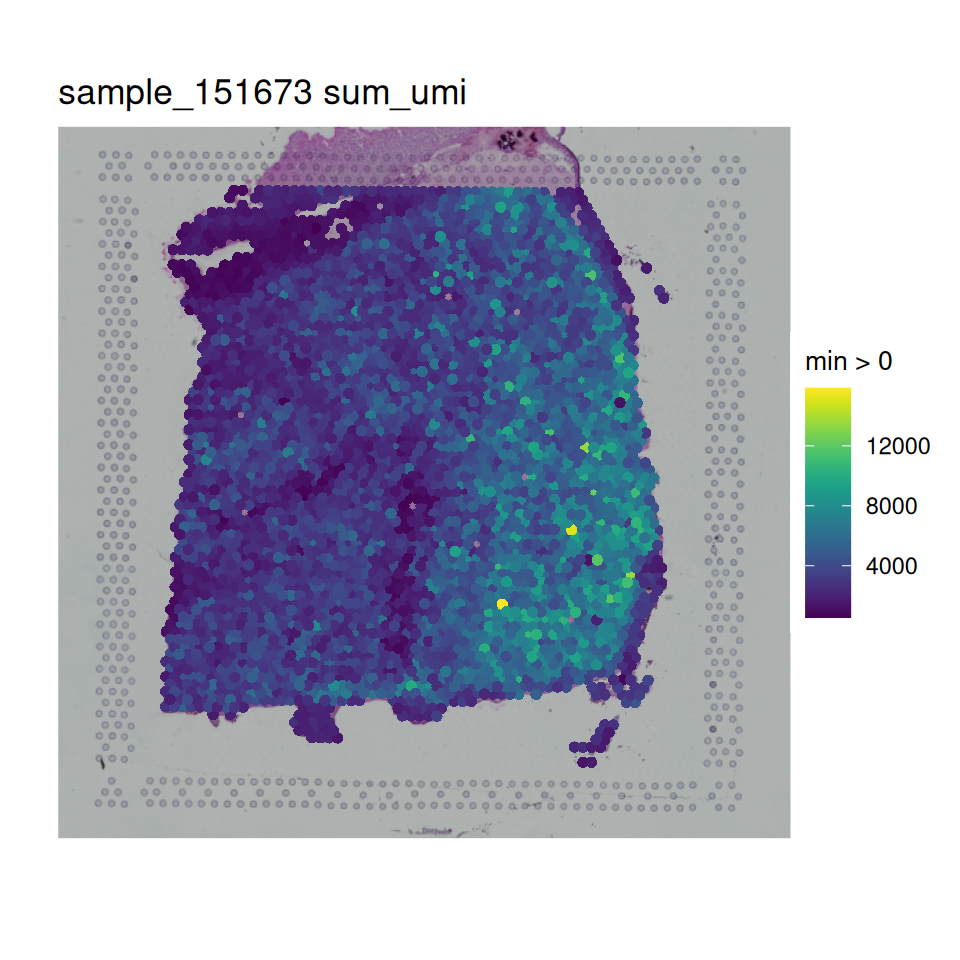
In order to visualize the data, we can then use spatialLIBD::vis_gene(). This is also a useful final check before we try launching our interactive website.
Code
# sum of UMI
spatialLIBD::vis_gene(
spe = spe,
sampleid = "sample_151673",
geneid = "sum_umi"
)
Code
Now, let’s proceed to visualize the data interactively with a spatialLIBD-powered website. We have a number of variables to choose from. We will specify which are the continuous and discrete variables in our spatialLIBD::run_app() call.
Code
# explore all the variables we can use
colData(spe)## DataFrame with 3614 rows and 26 columns
## barcode_id sample_id in_tissue array_row
## <character> <character> <integer> <integer>
## AAACAAGTATCTCCCA-1 AAACAAGTATCTCCCA-1 sample_151673 1 50
## AAACAATCTACTAGCA-1 AAACAATCTACTAGCA-1 sample_151673 1 3
## AAACACCAATAACTGC-1 AAACACCAATAACTGC-1 sample_151673 1 59
## AAACAGAGCGACTCCT-1 AAACAGAGCGACTCCT-1 sample_151673 1 14
## AAACAGCTTTCAGAAG-1 AAACAGCTTTCAGAAG-1 sample_151673 1 43
## ... ... ... ... ...
## TTGTTTCACATCCAGG-1 TTGTTTCACATCCAGG-1 sample_151673 1 58
## TTGTTTCATTAGTCTA-1 TTGTTTCATTAGTCTA-1 sample_151673 1 60
## TTGTTTCCATACAACT-1 TTGTTTCCATACAACT-1 sample_151673 1 45
## TTGTTTGTATTACACG-1 TTGTTTGTATTACACG-1 sample_151673 1 73
## TTGTTTGTGTAAATTC-1 TTGTTTGTGTAAATTC-1 sample_151673 1 7
## array_col ground_truth reference cell_count sum
## <integer> <character> <character> <integer> <numeric>
## AAACAAGTATCTCCCA-1 102 Layer3 Layer3 6 8458
## AAACAATCTACTAGCA-1 43 Layer1 Layer1 16 1667
## AAACACCAATAACTGC-1 19 WM WM 5 3769
## AAACAGAGCGACTCCT-1 94 Layer3 Layer3 2 5433
## AAACAGCTTTCAGAAG-1 9 Layer5 Layer5 4 4278
## ... ... ... ... ... ...
## TTGTTTCACATCCAGG-1 42 WM WM 3 4324
## TTGTTTCATTAGTCTA-1 30 WM WM 4 2761
## TTGTTTCCATACAACT-1 27 Layer6 Layer6 3 2322
## TTGTTTGTATTACACG-1 41 WM WM 16 2331
## TTGTTTGTGTAAATTC-1 51 Layer2 Layer2 5 6281
## detected subsets_mito_sum subsets_mito_detected
## <integer> <numeric> <integer>
## AAACAAGTATCTCCCA-1 3586 1407 13
## AAACAATCTACTAGCA-1 1150 204 11
## AAACACCAATAACTGC-1 1960 430 13
## AAACAGAGCGACTCCT-1 2424 1316 13
## AAACAGCTTTCAGAAG-1 2264 651 12
## ... ... ... ...
## TTGTTTCACATCCAGG-1 2170 370 12
## TTGTTTCATTAGTCTA-1 1560 314 12
## TTGTTTCCATACAACT-1 1343 476 13
## TTGTTTGTATTACACG-1 1420 308 12
## TTGTTTGTGTAAATTC-1 2927 991 13
## subsets_mito_percent total qc_lib_size qc_detected
## <numeric> <numeric> <logical> <logical>
## AAACAAGTATCTCCCA-1 16.6351 8458 FALSE FALSE
## AAACAATCTACTAGCA-1 12.2376 1667 FALSE FALSE
## AAACACCAATAACTGC-1 11.4089 3769 FALSE FALSE
## AAACAGAGCGACTCCT-1 24.2223 5433 FALSE FALSE
## AAACAGCTTTCAGAAG-1 15.2174 4278 FALSE FALSE
## ... ... ... ... ...
## TTGTTTCACATCCAGG-1 8.55689 4324 FALSE FALSE
## TTGTTTCATTAGTCTA-1 11.37269 2761 FALSE FALSE
## TTGTTTCCATACAACT-1 20.49957 2322 FALSE FALSE
## TTGTTTGTATTACACG-1 13.21321 2331 FALSE FALSE
## TTGTTTGTGTAAATTC-1 15.77774 6281 FALSE FALSE
## qc_mito discard sizeFactor label
## <logical> <logical> <numeric> <factor>
## AAACAAGTATCTCCCA-1 FALSE FALSE 1.834415 4
## AAACAATCTACTAGCA-1 FALSE FALSE 0.361548 6
## AAACACCAATAACTGC-1 FALSE FALSE 0.817440 1
## AAACAGAGCGACTCCT-1 FALSE FALSE 1.178338 2
## AAACAGCTTTCAGAAG-1 FALSE FALSE 0.927835 4
## ... ... ... ... ...
## TTGTTTCACATCCAGG-1 FALSE FALSE 0.937812 1
## TTGTTTCATTAGTCTA-1 FALSE FALSE 0.598820 1
## TTGTTTCCATACAACT-1 FALSE FALSE 0.503608 5
## TTGTTTGTATTACACG-1 FALSE FALSE 0.505559 7
## TTGTTTGTGTAAATTC-1 FALSE FALSE 1.362256 2
## key sum_umi sum_gene expr_chrM
## <character> <numeric> <integer> <numeric>
## AAACAAGTATCTCCCA-1 sample_151673_AAACAA.. 7051 3573 0
## AAACAATCTACTAGCA-1 sample_151673_AAACAA.. 1463 1139 0
## AAACACCAATAACTGC-1 sample_151673_AAACAC.. 3339 1947 0
## AAACAGAGCGACTCCT-1 sample_151673_AAACAG.. 4117 2411 0
## AAACAGCTTTCAGAAG-1 sample_151673_AAACAG.. 3627 2252 0
## ... ... ... ... ...
## TTGTTTCACATCCAGG-1 sample_151673_TTGTTT.. 3954 2158 0
## TTGTTTCATTAGTCTA-1 sample_151673_TTGTTT.. 2447 1548 0
## TTGTTTCCATACAACT-1 sample_151673_TTGTTT.. 1846 1330 0
## TTGTTTGTATTACACG-1 sample_151673_TTGTTT.. 2023 1408 0
## TTGTTTGTGTAAATTC-1 sample_151673_TTGTTT.. 5290 2914 0
## expr_chrM_ratio ManualAnnotation
## <numeric> <character>
## AAACAAGTATCTCCCA-1 0 NA
## AAACAATCTACTAGCA-1 0 NA
## AAACACCAATAACTGC-1 0 NA
## AAACAGAGCGACTCCT-1 0 NA
## AAACAGCTTTCAGAAG-1 0 NA
## ... ... ...
## TTGTTTCACATCCAGG-1 0 NA
## TTGTTTCATTAGTCTA-1 0 NA
## TTGTTTCCATACAACT-1 0 NA
## TTGTTTGTATTACACG-1 0 NA
## TTGTTTGTGTAAATTC-1 0 NACode
# run our shiny app
if (interactive()) {
spatialLIBD::run_app(
spe,
sce_layer = NULL,
modeling_results = NULL,
sig_genes = NULL,
title = "OSTA spatialLIBD workflow example",
spe_discrete_vars = c("ground_truth", "label", "ManualAnnotation"),
spe_continuous_vars = c(
"cell_count",
"sum_umi",
"sum_gene",
"expr_chrM",
"expr_chrM_ratio",
"sum",
"detected",
"subsets_mito_sum",
"subsets_mito_detected",
"subsets_mito_percent",
"total",
"sizeFactor"
),
default_cluster = "label"
)
}
12.3.6 Sharing your website
Now that you have created a spatialLIBD-powered website, you might be interested in sharing it. To do so, it will be useful to save a small spe object using saveRDS(), containing the data to share. The smaller the object, the better in terms of performance. For example, you may want to remove lowly expressed genes to save space. You can check the object size in R with object.size().
Code
# object size
format(object.size(spe), units = "MB")## [1] "308.1 Mb"If your data is small enough, you might want to share your website by hosting on shinyapps.io by Posit (the company that develops RStudio), which you can try for free. Once you have created your account, you need to create an app.R file like the one we have on the spatialLIBD_demo directory.
## library("spatialLIBD")
## library("markdown") # for shinyapps.io
##
## ## spatialLIBD uses golem
## options("golem.app.prod" = TRUE)
##
## ## You need this to enable shinyapps to install Bioconductor packages
## options(repos = BiocManager::repositories())
##
## ## Load the data
## spe <- readRDS("spe_workflow_Visium_spatialLIBD.rds")
##
## ## Deploy the website
## spatialLIBD::run_app(
## spe,
## sce_layer = NULL,
## modeling_results = NULL,
## sig_genes = NULL,
## title = "OSTA spatialLIBD workflow example",
## spe_discrete_vars = c("ground_truth", "label", "ManualAnnotation"),
## spe_continuous_vars = c(
## "cell_count",
## "sum_umi",
## "sum_gene",
## "expr_chrM",
## "expr_chrM_ratio",
## "sum",
## "detected",
## "subsets_mito_sum",
## "subsets_mito_detected",
## "subsets_mito_percent",
## "total",
## "sizeFactor"
## ),
## default_cluster = "label",
## docs_path = "www"
## )You can then open R in a new session in the same directory where you saved the app.R file, run the code and click on the “publish” blue button at the top right of your RStudio window. You will then need to upload the app.R file, your .rds file containing the spe object, and the files under the www directory which enable you to customize your spatialLIBD website.

The RStudio prompts will guide you along the process for authenticating to your shinyapps.io account, which will involve copy-pasting some code that starts with rsconnect::setAccountInfo(). Alternatively, you can create a deploy.R script and write the code for uploading your files to shinyapps.io as shown below.
## library('rsconnect')
##
## ## Or you can go to your shinyapps.io account and copy this
## ## Here we do this to keep our information hidden.
## # load(".deploy_info.Rdata")
## # rsconnect::setAccountInfo(
## # name = deploy_info$name,
## # token = deploy_info$token,
## # secret = deploy_info$secret
## # )
##
## ## You need this to enable shinyapps to install Bioconductor packages
## options(repos = BiocManager::repositories())
##
## ## Deploy the app, that is, upload it to shinyapps.io
## rsconnect::deployApp(
## appFiles = c(
## "app.R",
## "spe_workflow_Visium_spatialLIBD.rds",
## dir("www", full.names = TRUE)
## ),
## appName = 'OSTA_spatialLIBD_demo',
## account = 'libd',
## server = 'shinyapps.io'
## )Note that we have copied the default www directory files from the spatialLIBD repository and adapted them. We then use these files with spatialLIBD::run_app(docs_path) in our app.R script. These files help us control portions of our spatialLIBD-powered website and customize it.
If you follow this workflow, you will end up with a website just like this one. In our case, we further configured our website through the shinyapps.io dashboard. We selected the following options:
-
General
Instance Size: 3X-Large (8GB) -
Advanced
Max Worker Processes: 1 -
Advanced
Max Connections: 15
The Max Worker Processes determines how many R sessions are open per instance. Then Max Connections specifies the number of connections to each R session. The Instance Size determines the memory available. In this case, 8000 / 300 is approximately 27, but we decided to be conservative and set the total number of users per instance to 15. This is why it can be important to reduce the size of your spe object before sharing the website. Alternatively, you can rent an AWS Instance and deploy your app there, which is how http://spatial.libd.org/spatialLIBD is hosted along with these error configuration files.
12.3.7 Wrapping up
Thank you for reading this far! In this section we showed you:
- why you might be interested in using
spatialLIBD, - we re-used the
speobject from the DLPFC workflow (Section 12.2), - we adapted the
speobject to make it compatible withspatialLIBD, - we created an interactive website on our laptops,
- we shared the website with others using
shinyapps.io.