
17 Workflow: Visium HD (segmented)
17.1 Preamble
17.1.1 Introduction
As of June 2025, Visium HD enables direct output of H&E-segmented cell-level data, in addition to the previous binned_outputs at 2, 8, and 16 µm, from Space Ranger v4. This update simplifies the additional cell segmentation required when using bin2cell (Polański et al. 2024) or stardist (Weigert and Schmidt 2022). However, a comparative study of segmentation methods on Visium HD data has yet to be conducted. The morphology-driven, nucleus-based segmentation produces polygons representing nuclei and cells across the entire tissue, as illustrated in the schematics provided by 10x Genomics.
An alternative method, SMURF, “introduces the concept of soft segmentation, in which mRNA counts within a capture spot can be assigned wholly to a single cell or fractionally distributed among multiple nearby cells in a manner that optimizes the cosine similarity of each cell to its corresponding gene-expression cluster” (Guo et al. 2025).
17.1.2 Dependencies
Code
library(sf)
library(arrow)
library(dplyr)
library(tidyr)
library(scrapper)
library(SingleR)
library(Voyager)
library(ggplot2)
library(ggspavis)
library(VisiumIO)
library(OSTA.data)
library(patchwork)
library(SpotSweeper)
library(DropletUtils)
library(SpatialExperiment)
library(SpatialFeatureExperiment)
# set seed for random number generation
# in order to make results reproducible
set.seed(777)Code
# utility for cropping by bounding box
.crop <- \(spe, box) {
box <- as.list(box)
xy <- spatialCoords(spe)
spe[,
xy[,1] > box$xmin & xy[,1] < box$xmax &
xy[,2] > box$ymin & xy[,2] < box$ymax ]
}17.2 Setup
17.2.1 From SPE…
Analogous to other chapters, we start out by retrieving data files from the OSF repository using OSTA.data. Output files from Space Ranger v4 are located under /segmented_outputs (see Chapter 6 for details on Visium HD output structure). We can read these cell-level data into R as a SpatialExperiment (Righelli et al. 2022) using VisiumIO’s TENxVisiumHD() function by specifying the segmented_outputs argument accordingly.
Code
# retrieve data from OSF repo
id <- "VisiumHD_HumanColon_Oliveira"
pa <- OSTA.data_load(id)
dir.create(td <- tempfile())
unzip(pa, exdir=td)
# read into 'SpatialExperiment'
seg <- file.path(td, "segmented_outputs")
spe <- TENxVisiumHD(
format="h5",
images="lowres",
segmented_outputs=seg) |>
import()
# make gene symbols unique
gs <- rowData(spe)$Symbol
rownames(spe) <- make.unique(gs)
# needed for 'ggspavis'
spe$in_tissue <- TRUESpace Ranger also provides information on the mapping between binned outputs (2, 8, and 16 µm) and segmented cells. These are stored as cell metadata slot map:
Code
# view bin-to-cell mapping information
head(spe$map[[1]])## # A tibble: 6 × 6
## square_002um square_008um square_016um cell_id in_nucleus
## <chr> <chr> <chr> <chr> <lgl>
## 1 s_002um_02526_0… s_008um_00631_… s_016um_00315_… cellid_0000000… FALSE
## 2 s_002um_02526_0… s_008um_00631_… s_016um_00315_… cellid_0000000… FALSE
## 3 s_002um_02527_0… s_008um_00631_… s_016um_00315_… cellid_0000000… FALSE
## 4 s_002um_02527_0… s_008um_00631_… s_016um_00315_… cellid_0000000… TRUE
## 5 s_002um_02527_0… s_008um_00631_… s_016um_00315_… cellid_0000000… TRUE
## 6 s_002um_02527_0… s_008um_00631_… s_016um_00315_… cellid_0000000… TRUE
## # ℹ 1 more variable: in_cell <lgl>Quick inspection shows us the number of 2 µm bins that were segmented, either as part of nuclei or whole cells:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 1.00 15.00 24.00 28.55 38.00 185.00## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.02041 0.19048 0.30435 0.33725 0.45455 1.00000For runtime reasons, we will crop the tissue to about half of its original width and height.
Code
# reverse y-coordinates of bounding box
.box <- box
.box$ymax <- -box$ymin
.box$ymin <- -box$ymax
aes <- list(col="red", fill=NA, linewidth=2)
plotCoords(
spe[, sample(ncol(spe), 5e4)]) +
do.call(geom_rect, c(.box, aes)) +
plotCoords(
sub[, sample(ncol(sub), 5e4)])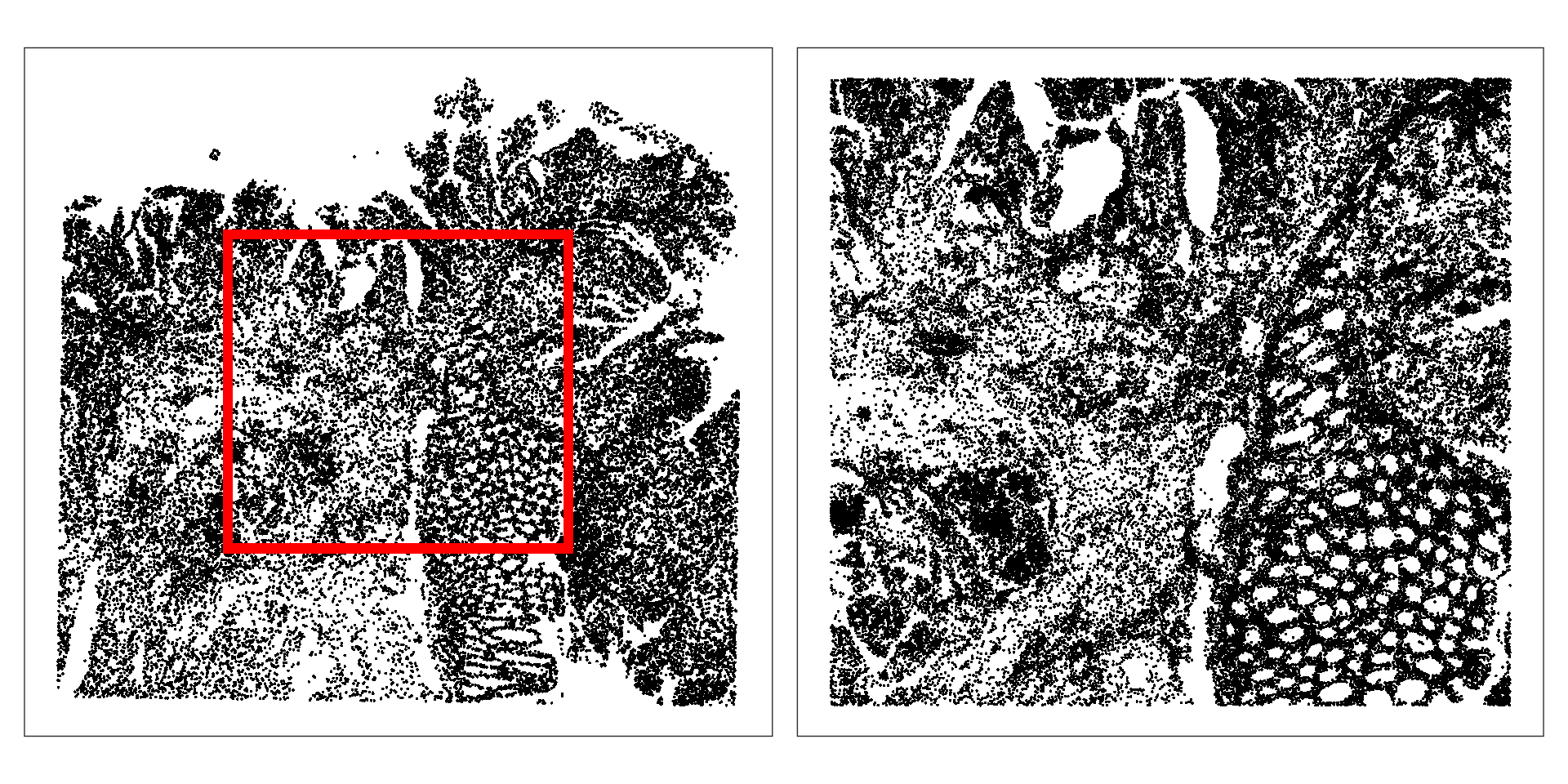
This retains about 28.55% of cells, and 63005 in total.
We also specify a much smaller region of interest (ROI) for visualization purposes.

17.2.2 …to SFE
Next, we will convert the above SPE into a SpatialFeatureExperiment (Moses et al. 2023) that allows us to also store cell segmentation masks as a colGeometry.
Code
# convert from SPE to SFE
sfe <- toSpatialFeatureExperiment(spe)
# add cell segmentation boundaries
seg <- metadata(spe)$cellseg
i <- colnames(spe)
j <- match(i, seg$cell_id)
seg <- seg[j, ]; rownames(seg) <- i
colGeometries(sfe) <- list(cellseg=seg)17.3 Exploratory
17.3.1 Cell masks
We can visualize such geometries using plot(st_geometry(x)) where x is the colGeometry of interest. As 63005 cells are a lot to visualize this way, we here crop() the data to filter for cells that fall within a small bounding box:
Code
# specify bounding box
box <- c(
xmin=56e3, xmax=58e3,
ymin=15e3, ymax=16e3)
# plot exemplary cell masks
geo <- colGeometry(crop(sfe, box))
plot(st_geometry(geo), col=rep(colors(), 2))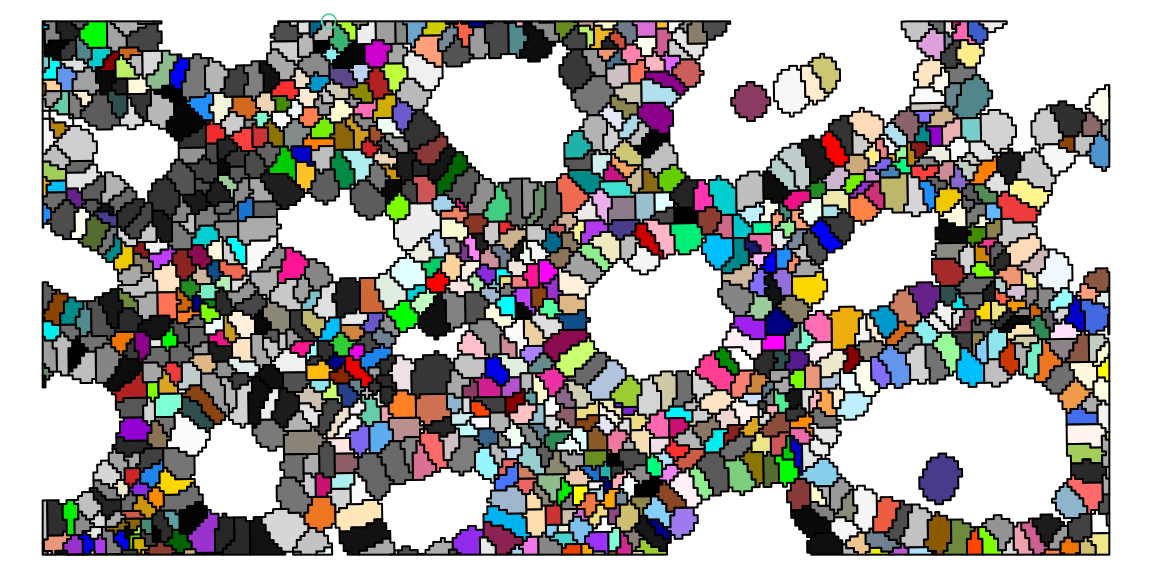
17.3.2 Bin mapping
We can estimate cell areas based on the number of 2 µm bins covered by each mask, e.g., 10 bins (4 µm2 each) would correspond to an area of 40 µm2. Based on this, we can also approximate the area of each cell’s nucleus using the in_nucleus flag provided in the bin-to-cell mapping information.
We can also investigate the number of different cells that are contained in 8 µm and 16 µm bins, respectively:
Code
## square_008um square_016um
## Min. 1.000000 1.000000
## 1st Qu. 1.000000 3.000000
## Median 2.000000 4.000000
## Mean 2.257041 4.513308
## 3rd Qu. 3.000000 6.000000
## Max. 8.000000 17.000000Here, we can observe that 8 µm/16 µm bins map to a median of 2/4 cells, only 25% of bins map to more than 3/6 cells, and at most 8/17 cells are mapped to any bin.
17.4 Quality control
17.4.1 Non-spatial
We can calculate standard cell-level quality control metrics using scrapper’s quickRnaQc.se() function, specifying mitochondrial features as subsets of particular interest.
Code
## DataFrame with 6 rows and 5 columns
## um2 nuc_um2 nuc sum
## <numeric> <numeric> <numeric> <numeric>
## cellid_000028786-1 52 16 0.307692 302
## cellid_000028793-1 152 28 0.184211 290
## cellid_000028794-1 76 28 0.368421 828
## cellid_000028795-1 156 40 0.256410 1121
## cellid_000028796-1 60 36 0.600000 1437
## cellid_000028799-1 96 20 0.208333 488
## subset.proportion.mt
## <numeric>
## cellid_000028786-1 0.0231788
## cellid_000028793-1 0.0448276
## cellid_000028794-1 0.0386473
## cellid_000028795-1 0.0428189
## cellid_000028796-1 0.0723730
## cellid_000028799-1 0.0204918Code
plotSpatialFeature(sfe[, sfe$roi],
colGeometryName="cellseg",
features="log_sum") +
ggtitle("log-library size") +
plotSpatialFeature(sfe[, sfe$roi],
colGeometryName="cellseg",
features="subset.proportion.mt") +
ggtitle("mitochondrial proportion") +
plot_layout(nrow=1) &
theme(plot.title=element_text(hjust=0.5)) &
scale_fill_gradientn(NULL, colors=pals::jet())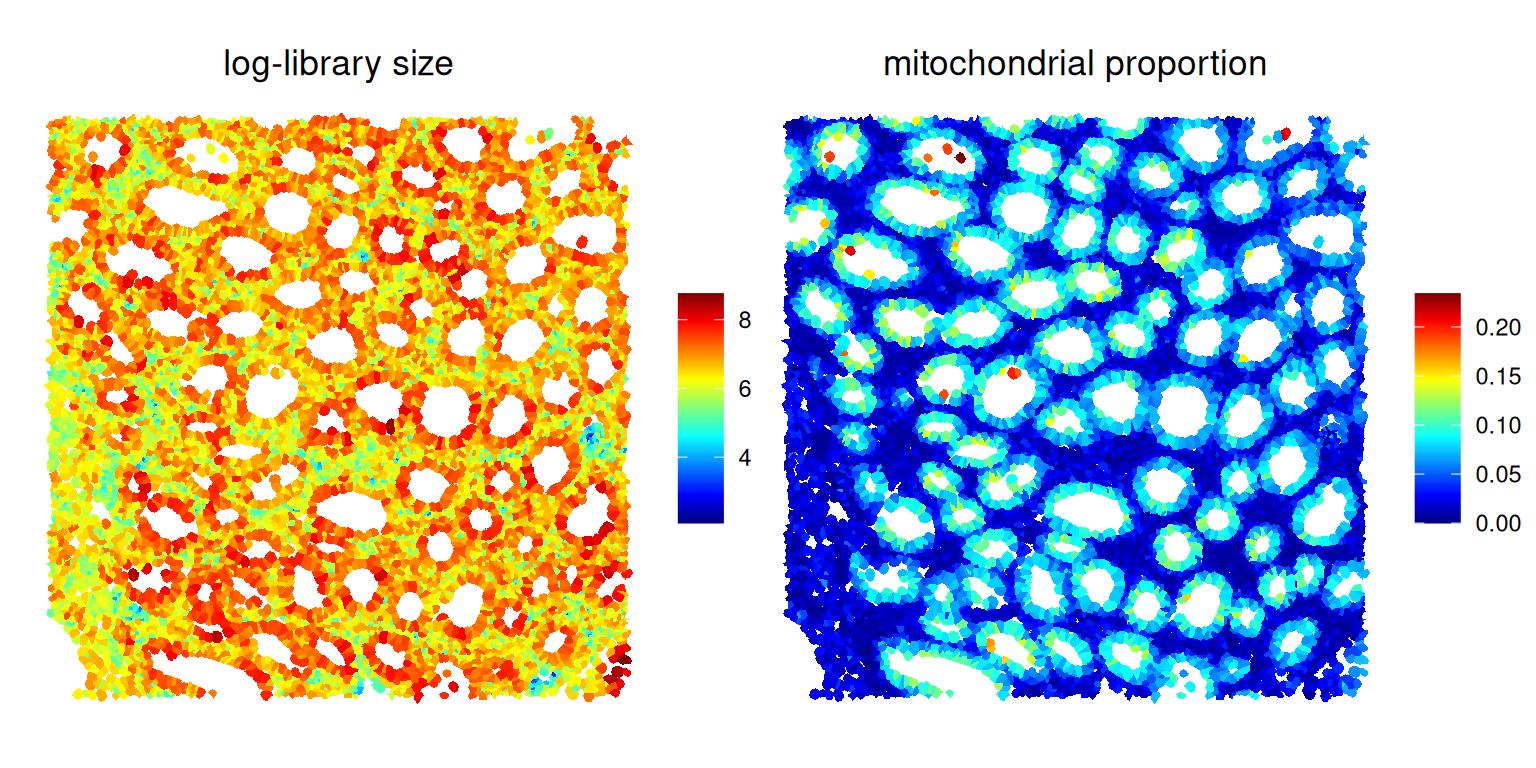
17.4.2 Spatially-aware
SpotSweeper (Totty et al. 2025) determines outliers based on low log-library size, few uniquely detected features, or a high mitochondrial fraction compared to their surrounding neighborhood, which we recommend (see Chapter 11).
Code
# determine spatial outliers for different metrics
sfe <- localOutliers(sfe,
workers=4, metric="sum",
log=TRUE, direction="lower")
sfe <- localOutliers(sfe,
workers=4, metric="detected",
log=TRUE, direction="lower")
sfe <- localOutliers(sfe,
workers=4, metric="subset.proportion.mt",
log=FALSE, direction="higher")
# get cells x flags matrix
cd <- colData(sfe)
ol <- grep("outliers$", names(cd))
ol <- as.matrix(cd[ol])
# percentage of cells excluded for each
round(100*colMeans(ol), 2)## sum_outliers detected_outliers
## 0.15 0.17
## subset.proportion.mt_outliers
## 0.44Code
# number of cells kept/removed overall
table(sfe$ex <- rowAnys(ol))##
## FALSE TRUE
## 62597 408We can visualize local outliers identified by SpotSweeper spatially. However, these are difficult to see, as only small cells were flagged.
Code
plotCoords(sfe[, order(sfe$ex)],
annotate="ex", point_size=0.2) +
theme(legend.key.size=ggplot2::unit(0, "pt")) +
scale_color_manual(values=c("grey90", "red")) +
guides(col=guide_legend(override.aes=list(size=2)))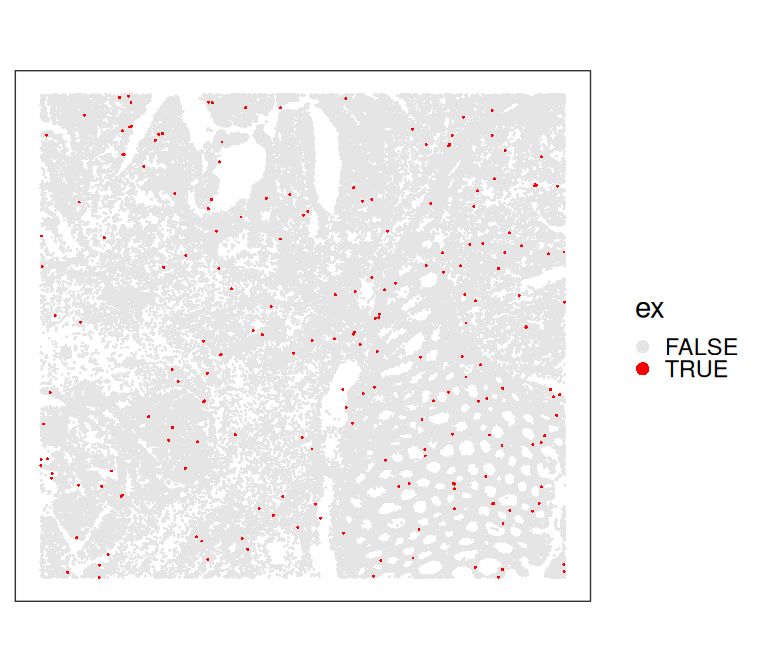
Code
# apply filtering
sfe <- sfe[, !sfe$ex]17.5 Annotation
This Visium HD dataset provides matched scRNA-seq for the same tissue, which avoids tissue and batch effects that often complicate label transfer. colData slot Level1 contains broad, interpretable annotations that are well suited for label-transfer, deconvolution, and downstream interpretation. We will use these to map likely cell identities onto our Visium HD cells.
Noteusing deconvolution instead
Here, we use a single-cell label transfer approach to annotate cells. However, although each observation is intended to represent a single cell, there are still cases where this is not the case (e.g., as a result of partial volume effects at boundaries, segmentation errors, tightly packed regions, or local diffusion of transcripts).
One could instead deconvolve cells (see Chapter 13), for example, using spacexr’s RCTD (Cable et al. 2022) in doublet mode to flag these cases and provide a principled estimate of the most likely composition. In practice, we expect most units to be classified as singlets, with a minority of doublets that warrant closer inspection or additional filtering.
Code
# retrieve single-cell reference from OSF repo
id <- "Chromium_HumanColon_Oliveira"
pa <- OSTA.data_load(id)
dir.create(td <- tempfile())
unzip(pa, exdir=td)
# read into 'SingleCellExperiment'
fs <- list.files(td, full.names=TRUE)
h5 <- grep("h5$", fs, value=TRUE)
sce <- read10xCounts(h5, col.names=TRUE)
# add cell metadata
csv <- grep("csv$", fs, value=TRUE)
cd <- read.csv(csv, row.names=1)
colData(sce)[names(cd)] <- cd[colnames(sce), ]
# use gene symbols as feature names
rownames(sce) <- make.unique(rowData(sce)$Symbol)
# exclude cells deemed to be of low-quality
sce <- sce[, sce$QCFilter == "Keep"]
# replace problematic characters
table(sce$Level1 <- gsub("\\s", "\\.", sce$Level1))##
## B.cells Endothelial Fibroblast
## 33611 7969 28653
## Intestinal.Epithelial Myeloid Neuronal
## 22763 25105 4199
## Smooth.Muscle T.cells Tumor
## 43308 29272 65626For the purpose of this demo, we downsample to at most 1,000 cells per reference class. This preserves diversity within each class while keeping memory and runtime manageable.
Code
## [1] 9000We next run BiocStyle::Biocpkg("SingleR") using Level1 (low-resolution) annotations and with argument aggr.ref=TRUE, such that reference profiles will be aggregated (per cluster) prior to annotation, and every cell will be assigned the label of the best-fitting pseudo-bulk scRNA-seq reference profile.
Code
# log-library size normalization
sfe <- normalizeRnaCounts.se(sfe)
.sce <- normalizeRnaCounts.se(.sce)
# perform label transfer at the Visium HD cell-level,
# using pseudo-bulk Chromium profiles as reference
res <- SingleR(test=sfe, ref=.sce, labels=.sce$Level1, aggr.ref=TRUE)## Detected a SingleCellExperiment as the reference dataset, consider setting
## 'de.method = "t"' or "wilcox" in trainSingleR(). If you know better, this hint
## can be disabled with 'hint.sce=FALSE'.Code
sfe$Level1 <- factor(res$pruned.labels)Simplifying further, we can group cells into different compartments, namely, (malignant) tumor, immune, epithelial and stromal cells; we’ll see below that visualizing cells in this way nicely captures the general tissue structure.
Code
##
## epi imm str tum
## 6911 11934 15899 27716We can visualize low-resolution and compartment-level annotations over our ROI.
Code
pal_lv1 <- unname(pals::trubetskoy())
pal_lv0 <- c("gold", "turquoise", "deeppink", "navy")
plotSpatialFeature(sfe[, sfe$roi],
features="Level1", colGeometryName="cellseg") +
scale_fill_manual(values=pal_lv1) +
plotSpatialFeature(sfe[, sfe$roi],
features="Level0", colGeometryName="cellseg") +
scale_fill_manual(values=pal_lv0) +
plot_layout(nrow=1) &
theme(legend.key.size=ggplot2::unit(.5, "lines"))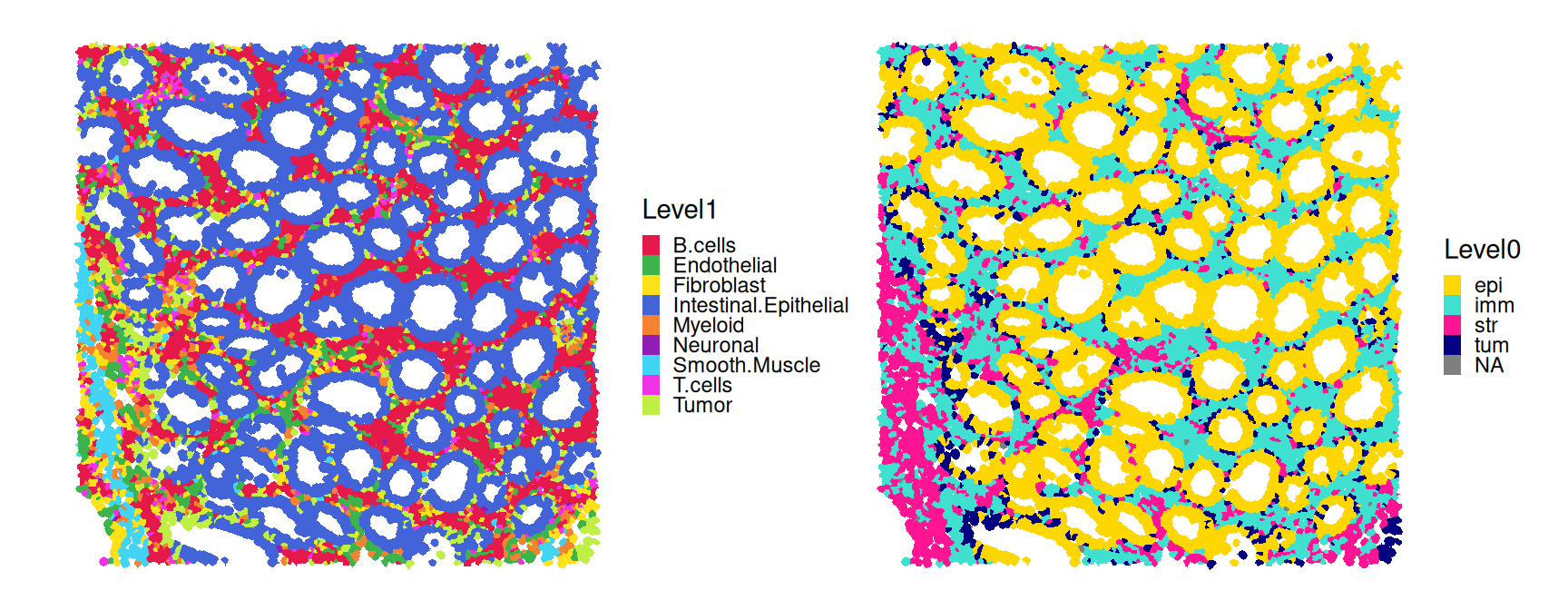
For whole-section visualization, we do not render segmentation boundaries but only cell centroids.
Code
plotCoords(sfe,
annotate="Level1", point_size=0.1) +
scale_color_manual(values=pal_lv1) +
plotCoords(sfe,
annotate="Level0", point_size=0.1) +
scale_color_manual(values=pal_lv0) +
plot_layout() &
theme(legend.key.size=ggplot2::unit(0, "pt"))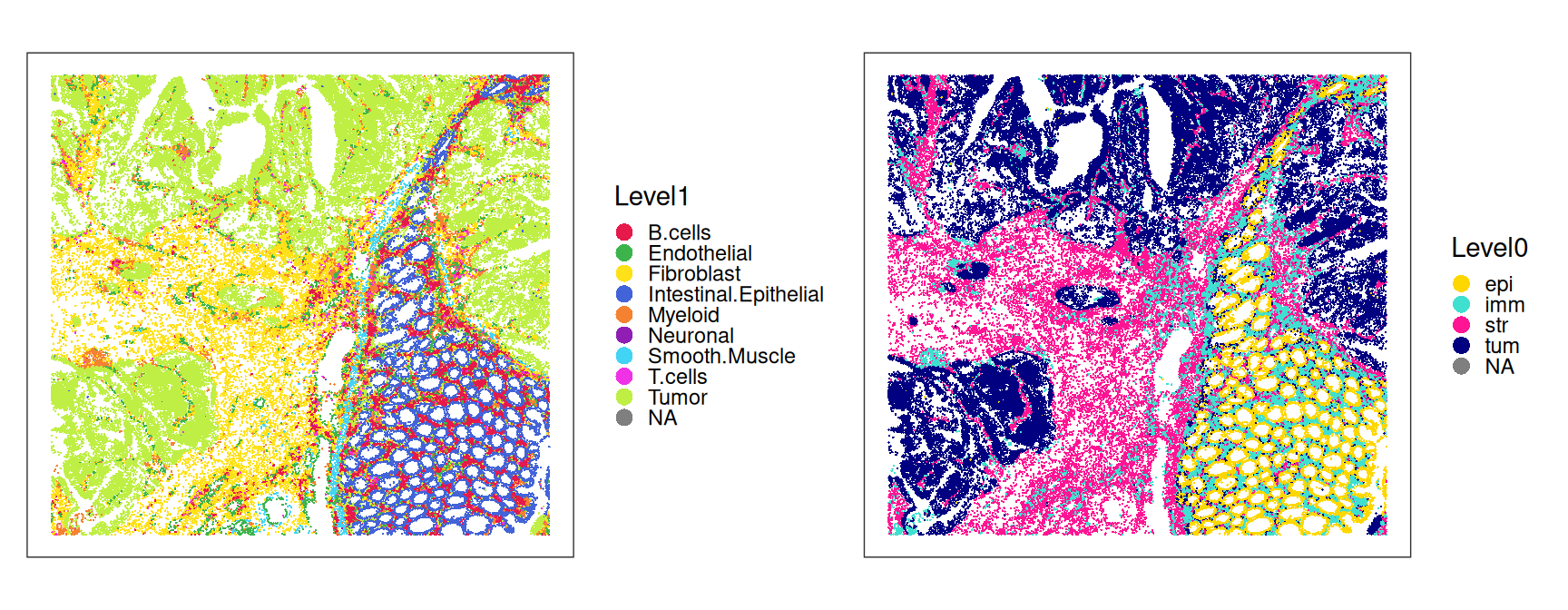
17.6 Downstream
Having annotated our cells, we can proceed with various downstream analyses. Compared to analysis at the bin-level, cell-level Visium HD data is similar to data from imaging-based platforms, but with full-transcriptome coverage. In principle, most analyses are thus transferable.
We refer readers to the chapters designed to specific analysis tasks, e.g., Chapter 30 on feature selection and testing, and Chapter 31 on feature-set scoring.
Instead, we quickly inspect how cell and nucleus areas differ across compartments and subpopulations. As expected, immune cells are smallest, tumor and structural cells (epithelial and stromal cells) tend to be larger.
Code
# wrangling
df <- data.frame(colData(sfe))
df <- filter(df, !is.na(Level0))
fd <- pivot_longer(df, ends_with("um2"))
fd$name <- factor(fd$name, labels=c("nuc.", "cell"))
ws <- c(nlevels(df$Level0), nlevels(df$Level1))
# plotting
ggplot(fd, aes(reorder(Level0, value), value, col=name)) +
ggplot(fd, aes(reorder(Level1, value), value, col=name)) +
plot_layout(nrow=1, widths=ws, guides="collect") &
geom_boxplot(key_glyph="point", outlier.size=0) &
labs(col="area (um2)") &
theme_bw() & theme(
axis.title=element_blank(),
legend.key.size=ggplot2::unit(0, "pt"),
axis.text.x=element_text(angle=45, hjust=1)) 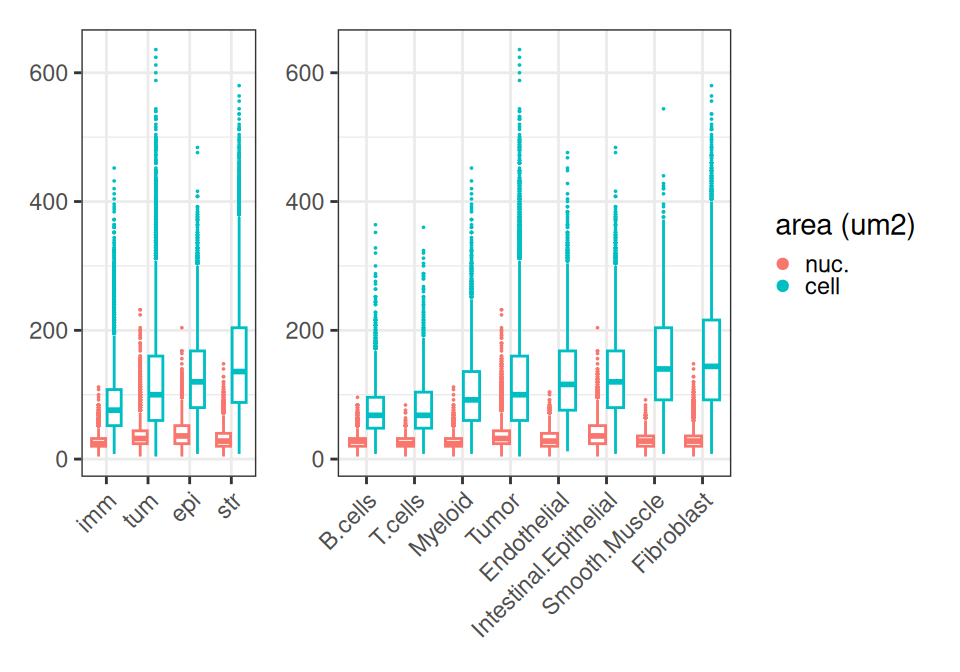
17.7 Appendix
References
Cable, Dylan M., Evan Murray, Luli S. Zou, et al. 2022. “Robust Decomposition of Cell Type Mixtures in Spatial Transcriptomics.” Nature Biotechnology 40: 517–26. https://doi.org/10.1038/s41587-021-00830-w.
Guo, Juanru, Simona Sarafinovska, Ryan A Hagenson, et al. 2025. “SMURF: soft-segmentation for single-cell reconstruction and topological analysis of spatial transcriptomic data.” bioRxiv, ahead of print. https://doi.org/10.1101/2025.05.28.656357.
Moses, Lambda, Pétur Helgi Einarsson, Kayla Jackson, et al. 2023. “Voyager: Exploratory Single-Cell Genomics Data Analysis with Geospatial Statistics.” bioRxiv, ahead of print. https://doi.org/10.1101/2023.07.20.549945.
Polański, Krzysztof, Raquel Bartolomé-Casado, Ioannis Sarropoulos, et al. 2024. “Bin2cell Reconstructs Cells from High Resolution Visium HD Data.” Bioinformatics 40 (btae546, 9). https://doi.org/10.1093/bioinformatics/btae546.
Righelli, Dario, Lukas M Weber, Helena L Crowell, et al. 2022. “SpatialExperiment: Infrastructure for Spatially-Resolved Transcriptomics Data in r Using Bioconductor.” Bioinformatics 38: 3128–31. https://doi.org/10.1093/bioinformatics/btac299.
Totty, Michael, Stephanie C. Hicks, and Boyi Guo. 2025. “SpotSweeper: Spatially Aware Quality Control for Spatial Transcriptomics.” Nature Methods 22: 1520–30. https://doi.org/10.1038/s41592-025-02713-3.
Weigert, Martin, and Uwe Schmidt. 2022. “Nuclei Instance Segmentation and Classification in Histopathology Images with Stardist.” The IEEE International Symposium on Biomedical Imaging Challenges (ISBIC). https://doi.org/10.1109/ISBIC56247.2022.9854534.