library(SpatialExperiment)
spe <- readRDS("spe_clustering.rds")13 Differential expression
13.1 Introduction
In this chapter, we perform differential expression testing between clusters or spatial domains to identify representative marker genes for each cluster or spatial domain.
13.2 Load previously saved data
We start by loading the previously saved data object(s) (see Section 10.4).
13.3 Differential expression testing
Identify representative marker genes for each cluster or spatial domain by testing for differential gene expression between clusters.
Here, we use the findMarkers implementation in scran (Lun, McCarthy, and Marioni 2016), using a binomial test, which tests for genes that differ in the proportion expressed vs. not expressed between clusters. This is a more stringent test than the default t-tests, and tends to select genes that are easier to interpret and validate experimentally.
# set gene names as row names for easier plotting
rownames(spe) <- rowData(spe)$gene_name
# test for marker genes
markers <- findMarkers(spe, test = "binom", direction = "up")
# returns a list with one DataFrame per cluster
markers## List of length 6
## names(6): 1 2 3 4 5 6# plot log-fold changes for one cluster over all other clusters
# selecting cluster 1
interesting <- markers[[1]]
best_set <- interesting[interesting$Top <= 5, ]
logFCs <- getMarkerEffects(best_set)
pheatmap(logFCs, breaks = seq(-5, 5, length.out = 101))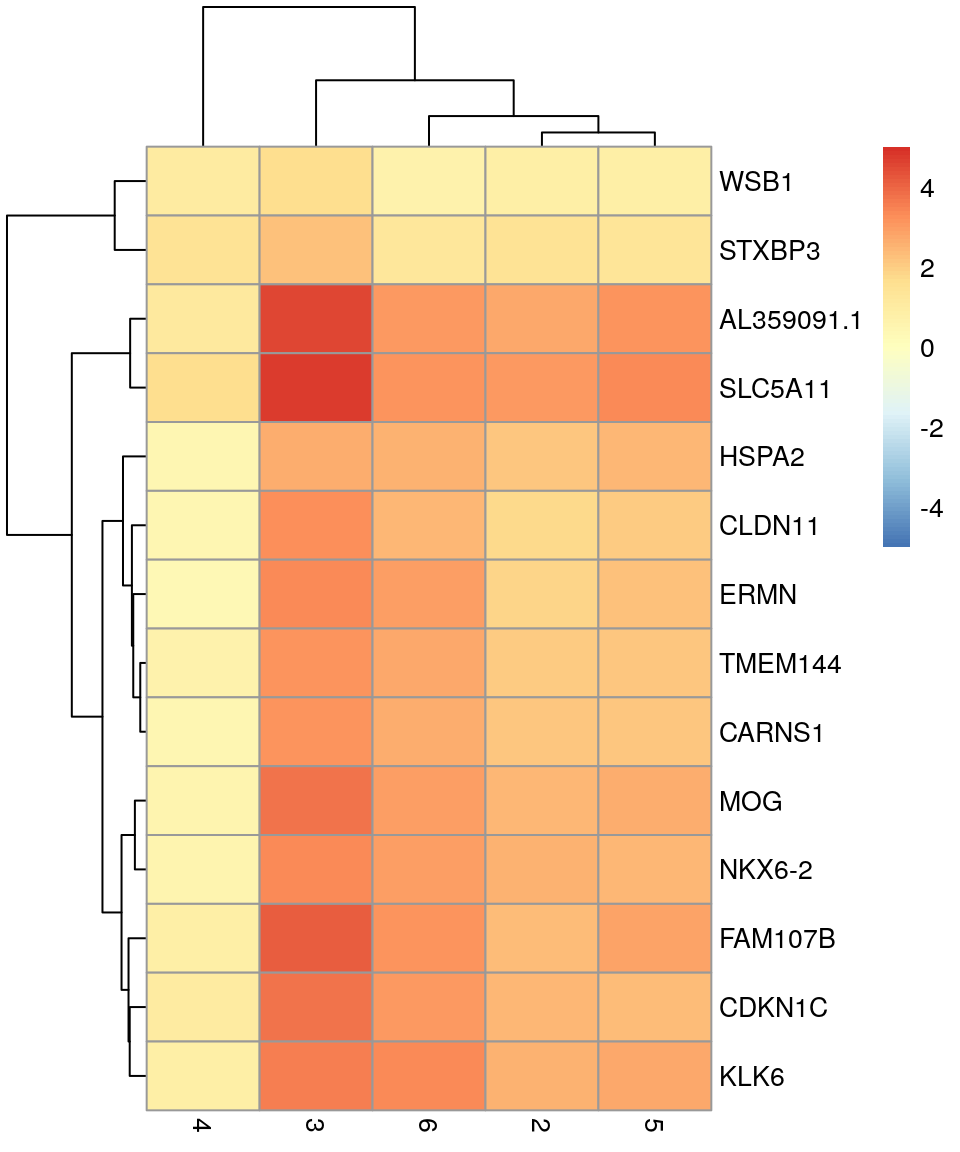
# plot log-transformed normalized expression of top genes for one cluster
top_genes <- head(rownames(interesting))
plotExpression(spe, x = "label", features = top_genes)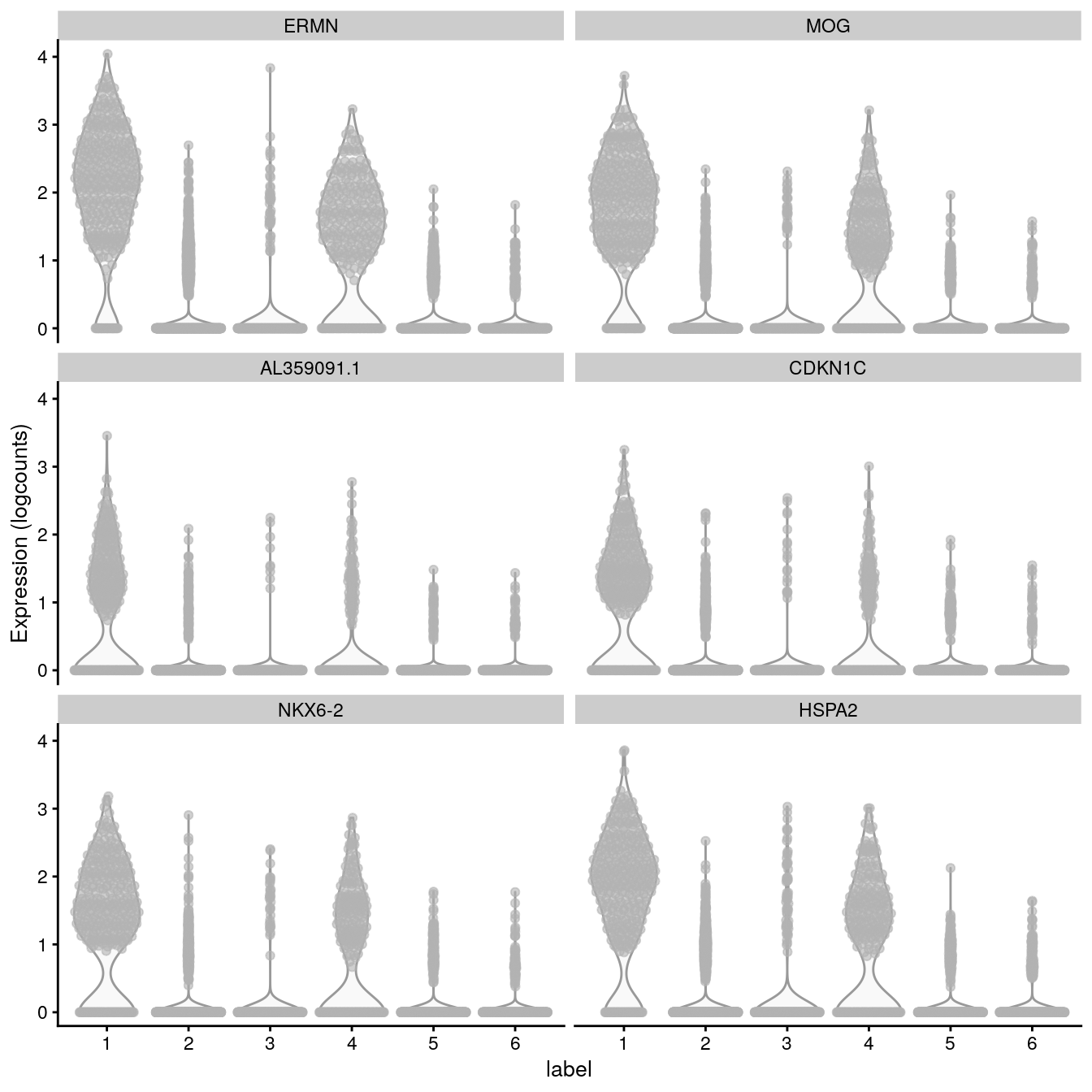
13.4 Pseudobulking
Alternatively, we can proceed by manually aggregating the counts per cluster or spatial domain, which is referred to as “pseudobulking”. Then, we can perform differential expression testing between the pseudobulked clusters or spatial domains.
13.5 Multiple samples
In the preceding chapters, we have focused on spatial transcriptomics datasets consisting of a single tissue section, which we refer to as a sample.
Datasets consisting of multiple samples from one or more biological conditions are used to study a variety of biological questions, such as characterizing biological variability within replicate tissue samples, or differential comparisons between biological conditions.
These datasets require additional analysis methods that can make full use of the information contained in multiple samples in a computationally efficient and statistically rigorous manner.
References
Lun, Aaron T. L., Davis J. McCarthy, and John C. Marioni. 2016. “A Step-by-Step Workflow for Low-Level Analysis of Single-Cell RNA-seq Data with Bioconductor.” F1000Research 5 (2122). https://doi.org/10.12688/f1000research.9501.2.